Abstract
Almost 30 years have passed since the first successful hematopoietic stem cell transplantation in thalassemia and that first patient is now a healthy young adult with a completely normal life. Since that time, more than 3000 such transplants have been performed worldwide. This review provides a brief history of hematopoietic stem cell transplantation in thalassemia and reassesses current clinical results with the objective to provide outcome predictions based on modern transplant technologies. The role of hematopoietic stem cell transplantation in the oral chelation era and implications for possible closure in the approach to future gene therapy will also be discussed.
Hematopoietic Stem Cell Transplantation in Thalassemia: A Model of Cellular Replacement Therapy
Allogeneic hematopoietic stem cell (HSC) transplantation (HSCT) in thalassemia has been a cornerstone in the development of HSCT.1,2 The rational basis of HSCT in thalassemia consists of substituting the thalassemic HSC bearing ineffective erythropoiesis with an allogeneic one capable of effective erythropoiesis. This cellular replacement therapy is not limited to the diseased erythropoietic component, but leads to the replacement of the entire hematopoietic system. Nevertheless, it is an efficient way to obtain a long-lasting, probably permanent, clinically effective correction of hemolytic anemia, thus avoiding transfusion requirements and associated complications (ie, iron overload).
The transplantation approach for a nonmalignant disease is much different from transplantation in malignancies. In the former setting, the detrimental immunologic proprieties (ie, graft-versus-host disease [GVHD]) of the engrafted HSC are not balanced by an antimalignancy effect. This characteristic must be always considered in determining the risk/benefit ratio and therapeutic decision such as the kind and intensity of the conditioning regimen, GVHD prophylaxis, source of HSC, and adoptive posttransplant therapies.
History of HSCT in Thalassemia
HSCT in thalassemia was developed and grew into accepted routine clinical practice primarily thanks to the Pesaro Group experience during the 1980s and early 1990s.3–9 During that period, more than 1000 unselected thalassemia patients were transplanted in Pesaro, with an overall 20 years thalassemia-free survival of 73% calculated on 900 consecutive patients (Figure 1), who received transplants from an HLA identical sibling.10 These results have been confirmed by the Pescara Group, who conducted a similar experience in more than 100 selected patients from the early 1980s11 (Figure 2).

Results of 900 consecutive unselected HSCTs for thalassemia performed in Pesaro since 1982. Reprinted with permission from Angelucci and Baronciani.10
Results of 900 consecutive unselected HSCTs for thalassemia performed in Pesaro since 1982. Reprinted with permission from Angelucci and Baronciani.10

Results of 115 consecutive HSCTs for thalassemia in Pescara since 1984. Reprinted with permission from Di Bartolomeo et al.11
Results of 115 consecutive HSCTs for thalassemia in Pescara since 1984. Reprinted with permission from Di Bartolomeo et al.11
Figure 3 shows the number of transplants for thalassemia referred to the European Group for Blood and Marrow Transplantation (EBMT). The hemoglobinopathy registry surpassed the 100 transplants per year threshold in 1989 and has varied from year 2000 on from a low of 129 per year to a high of 197 per year.

HSCTs in thalassemia by years. Data from the Hemoglobinopathies Registry of the European Group for Blood and Marrow Transplantation. Reprinted with permission from Angelucci and Baronciani.10
HSCTs in thalassemia by years. Data from the Hemoglobinopathies Registry of the European Group for Blood and Marrow Transplantation. Reprinted with permission from Angelucci and Baronciani.10
In the 1980s, the Pesaro group developed a prognostic scheme to predict transplant outcome in patients younger than age 17.6 This prognostic scheme included three variables all related to iron burden: (1) quality of chelation received for the entire life before transplantation; (2) hepatomegaly; and (3) the presence of liver fibrosis at pretransplant hepatic biopsy examination. These variables stratified patients into three groups based on them having either none, or one/two, or all three of the risk factors. Overall survival and thalassemia-free survival were significantly different in the three groups: 94% and 87% in the low-risk group, 84% and 81% in the intermediate-risk group, and 70% and 58% in the high-risk group, respectively.12 No similar results have been achieved in adult patients, with overall survival and thalassemia-free survival peaking at 67% to 63%, respectively, and a transplant mortality rate of 35%.12 The risk classification for pediatric patients was not applicable to adult patients,9 mostly likely because very few adult patients could have received complete regular chelation in the deferoxamine era.
The rate of rejection/thalassemia recurrence was much higher in thalassemic patients than in patients transplanted because of malignancies. Several factors have been invoked to explain this difference: massive pretransplant exposure to blood products, not having received any chemotherapy before transplant, expanded erythropoietic marrow, and possibly splenomegaly. Interestingly, the phenomenon of high thalassemia recurrence was limited to patients younger than age 17.
A major criticism of the Pesaro classification has been that two of the variables, chelation quality and hepatomegaly, are qualitative and subject to intra- and interobserver variabilities. This is a valid criticism and quantitatively defined risk factors based on defined units of measure would obviously increase the precision, accuracy, and applicability of the classification; however, so far, all attempts to identify such criteria have failed. The value of the Pesaro classification is that it provides a powerful demonstration of how the quality of medical care that patients have received during their entire life before transplant is a critical determinant of transplant outcome. Prevention of transfusion and iron overload-related tissue damage primarily yields a greater probability to overcome the toxic and immunologic insults that are required to obtain and sustain engraftment. Gaziev et al13 demonstrated, in a large retrospective study, that GVHD incidence and severity were similar in the three categories of patients, but what was much different was the probability of surviving GVHD: mortality rate following acute grade III/IV GVHD was 27%, 48%, and 84% for low-, intermediate-, and high-risk patients, respectively.
This demonstrated link between medical therapy and transplantation is a clear demonstration that these two approaches are mutually related. Optimal medical therapy is essential for successful transplant, and the prospective of a potential curative approach is important for optimal medical therapy. Moreover optimization of chelation status leading to transplant (much more feasible today because of recent advances in chelation) could at least partially reverse the demonstrated deleterious effects of iron overload.
The growing experiences in long-term follow-up after transplant further enhance the link between medical therapy and transplantation. The same relationship will most likely apply to future approaches to gene therapy.
Current Results
Transplant technologies and clinical care have widely improved during the last decade, with outstanding results being constantly reported in the literature. Several centers worldwide are now performing HSCT in thalassemia with excellent results.14 Various approaches have been developed for HSCT preparation and conditioning: intravenous busulfan14 ; targeted intravenous busulfan15 ; new drugs like treosulfan,16 thiotepa, and fludarabine,17 as well as intensive pretransplant transfusion-chelation regimens.
These different regimens have more or less yielded the same overall survival and thalassemia-free survival, which are now greater than 90% and more than 80%, respectively, in patients without relevant iron-related tissue damage. Similar results have been reported by a retrospective EBMT survey, including more than 1000 patients transplanted in the last decade in 120 centers worldwide (E. Angelucci, unpublished observations, 2010).
Survival increased at times above 90%, even in high-risk pediatric patients, but with thalassemia-free survival ranging from 66% to 80%14–16 probably because of the reduced toxicity regimen usually applied to this category of patients.
There is only a single report on a limited number of adult patients (n = 15) transplanted after 1997, with modest improvements in results (overall survival 67%; transplant-related mortality 27%).18
Alternative Source of HSC
The large majority of transplant centers continue to use bone marrow-derived HSC rather than peripheral blood-derived HSC. Young age of the sibling donors and the lack of a requirement for a graft versus malignancies effect are the two most likely reasons for the predominant use of bone marrow-derived HSC.
In 2003, Locatelli et al19 first reported feasibility of using HLA identical sibling, cord blood-derived HSC for HSCT in thalassemia. As expected, this kind of transplant was associated with a decreased risk of acute and chronic GVHD and transplant-related mortality. A large, ongoing international study—retrospectively comparing cord blood to bone marrow receivers (each matched for age, risk, and center experience) in thalassemia—has begun to yield comparable results with less GVHD occurrence.20 In this study, overall survival, thalassemia-free survival, acute GVHD, and chronic GVHD were 95%, 88%, 20%, and 12% for bone marrow recipients (n = 389, mean age = 8 years), respectively, and 96%, 81%, 10%, and 5% for cord blood recipients (n = 70, mean age = 6 years), respectively. Almost all patients included belonged to the Pesaro “good-risk” categories.
In this contest, it must be mentioned that the recent development of preimplantation diagnosis for a single gene disease and preimplantation HLA typing21 have made it possible to select a nonthalassemic HLA identical infant donor for an existing patient.22 Currently, this technique is feasible for thalassemia and other nonmalignant, and even some malignant, diseases. However, the procedure raises very important ethical, legal, and financial controversies. In some countries, such as Italy, embryo selection on the basis of HLA matching is not allowed.
Alternative Donors
Clinical development of HSCT from alternative donors has been more challenging. It includes 3 possible approaches: (1) matched unrelated donors, (2) mismatched related donors, and (3) unrelated cord blood.
Matched Unrelated Donors.
During the last decade, use of unrelated donors for HSCT in malignancies has expanded considerably with outstanding results. Crucial determinants of this success have been technological improvement of molecular HLA typing and improved capability of selecting the appropriate unrelated donor.
A multicenter GITMO (Gruppo Italiano Trapianto Midollo Osseo) study started more than 10 years ago recently presented 68 thalassemic patients transplanted from a matched unrelated donor.23 In this group of pediatric and adult patients (age range 2–37, median age 15 years), overall survival and thalassemia-free survival reached 79% and 66%, respectively. In the group of 30 pediatric patients in the two lower risk Pesaro categories, overall survival and thalassemia-free survival were 97% and 80%, respectively, whereas in the high-risk group, they were 65% and 54%, respectively. If stringent criteria of immunogenetic compatibility—based on high-resolution molecular typing—are respected, results are similar to those obtained in the matched sibling setting. It is crucial to achieve extended haplotype identity (ie, identity from locus HLA-A to locus HLA-Dq on the same chromosome). A recent study has demonstrated that the risk of thalassemia recurrence after unrelated bone marrow transplantation is associated with the presence of nonpermissive HLA-DPB1 mismatches in the host-versus-graft direction.24 In this experience, the prognostic significance of Pesaro risk stratification has also been confirmed.
The capability of selecting the best available unrelated donor is much improved today.25 However, in addition to costs, there are two major limitations to this otherwise successful approach: (1) the stringent matching criteria outlined previously strongly limit donor availability (data on search success/failure with those criteria are not available) and (2) unrelated donor registries are not well represented in ethnic group with a high incidence of thalassemia.
Mismatched Related Donors.
The experience in this setting has been limited so far, with encouraging, but still suboptimal results (probability of overall survival and thalassemia-free survival of 65% and 21%, respectively, with a median follow-up of 7 years in a consecutive series of 29 patients).26 Only recently, better results have been reported using an haploidentical related donor after positive selection of CD34+ cells in a small series (n = 22) of heterogeneous thalassemia patients.27 In this series, there were two deaths, six thalassemia recurrences, and 14 patients had sustained engraftment. However, this limited, single center experience does not support at the present time a wider use of this approach.
Unrelated Cord Blood.
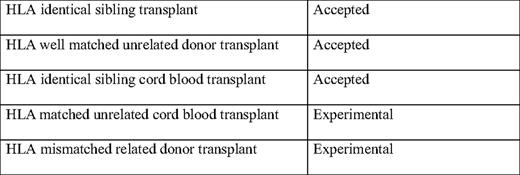
Unrelated cord blood transplantation has not been explored in systematic studies and only one single center study28 and retrospective multicenter studies are available. In the single center study from Taiwan on 32 low-risk patients, 27 were alive and transfusion-independent after a median follow-up of 27 months. Fifty-one such transplants have been reported in the registries at EBMT and CIBMTR (Center for International Blood and Marrow Transplant Research).29 Of these 51 patients, 13 have died and only 16 presented a sustained engraftment. Globally, 2-year overall survival was 77%. Based on the reported results, this approach cannot be recommended unless it is part of a controlled clinical trial. Table 1 provides a summary of the accepted and experimental HSCT approaches.
Nonmyeloablative Conditioning Regimens
Nonmyeloablative HSCT has the theoretical advantage, based on the experience with malignancies, of obtaining allogeneic engraftment with a very low, early mortality rate. However, increased reliance on immunologic effects that is required to sustain engraftment requires a prudent approach to the wide use of this regimen, and very few cases have been reported so far. Lucarelli and Gaziev14 reviewed the limited international experience in thalassemia and sickle cell disease that showed overall poor results and a very reduced rate of sustained engraftment (only 1 in 11 transplants). More recently, a successful trial has been published in a small cohort of adult patients with sickle cell disease (n = 11) conditioned with 300 cGy of total body irradiation and alemtuzumab.30 However, this study is not immediately applicable to thalassemia. There are, in fact, several relevant differences between thalassemia major and sickle cell disease, which impact HSCT-based approaches. Thalassemia major is characterized by ineffective erythropoiesis and variable erythroid expansion.31 Ineffective erythropoiesis and chronic transfusion lead to iron overload. Thus, for thalassemia, a conditioning regimen capable of eradicating an expanded bone marrow and providing adequate immunosuppression to sustain engraftment with acceptable toxicity on iron-damaged tissues is required. These challenges are not present in sickle cell disease. The approach to patients with sickle cell disease is different than in thalassemias, due to the fact that transfusion therapy is not standard practice for all patients, the transfusion burden is less relevant, and the expansion of the erythroid marrow is not as massive.
Mixed Chimerism
The Pesaro experience has shown that a significant group of patients (11% of the entire Pesaro experience) develop long-term, stable-mixed chimerism after transplantation.32 This has been confirmed by other groups.11 Mixed chimeric patients, despite a limited (even 20%) engraftment, achieve a functioning graft status characterized by normal hemoglobin level, no red blood cell transfusion requirement, no iron increment, and a limited—not clinically relevant—erythroid hyperplasia.31 Thus, in chimeric patients, the genetic disease is under a substantially complete clinical control, without achieving complete eradication of the thalassemic hemopoietic clones.33
Immunologically, mixed chimera patients had normal lymphoid subset distribution, normal response to mitogens stimulation, normal response to allogeneic antigens, and normal cytotoxicity activity. Three aspects of the mixed chimerism phenomenon require additional attention: (1) kinetic, (2) immunologic, and (3) erythroid.
Kinetic.
Andreani et al33 demonstrated that “early” (day 60 after HSC transplant) chimerism is a risk factor for thalassemia recurrence in patients receiving bone marrow-derived HSCs and that an “early” engraftment greater than 90% is required for a high probability to obtain stable either full or mixed chimerism. This finding can be interpreted as a requirement for a “bulk” engraftment effect that is necessary to achieve a sustained, long-term engraftment (with either full or persistent mixed chimerism). How to manage this situation or how to manage declining donor chimerism is still the subject of intense debate, because the risk/benefit ratio of adoptive immunotherapy in these circumstances is controversial and the impact of host recovery is not well understood.34,35
This “bulk engraftment effect” may explain the low success rate of the nonmyeloablative approach in the thalassemic population. The Locatelli Group demonstrated that this potential “bulk” effect does not seem to be necessary in HLA identical sibling cord blood transplants.36
Immunologic.
The underlying mechanisms leading to HSC coexistence have been the object of intense investigation, with no clear answer yet emerging. Conditioning, toxicity, GVHD, and other factors do not seem to play a role. Serafini et al37 showed that the alloreactive T-cell clones of both host and donor origins, isolated from the peripheral blood of the mixed chimera patients, could function as partially effector T cells reactive against host or donor alloantigens. Moreover, these cells were partially regulatory T cells with a cytokine secretion profile typical of type 1 regulatory T cells. Importantly, the regulatory T-cell clones, both donor and host derived, were able to inhibit cytokine production and proliferation of effector cells of either donor or host origin, suggesting a contribution by these regulatory T cells to the maintenance of persistent mixed chimerism in vivo. These data provide new insights into the mechanisms of peripheral tolerance and may pave the way for cellular therapy trials following HSC transplantation in nonmalignant diseases.
The presence of residual host cells may also contribute to reduce the incidence and severity of GVHD, as, in animal models, mixed chimera is associated with reduced susceptibility to GVHD, probably through a mechanism of central tolerance with negative selection of host-reactive donor T cells.38,39
The different mixed chimerism kinetics development reported in cord blood transplantation indicate that cord blood transplant is more effective than bone marrow transplant in promoting a state of reciprocal tolerance between recipient and donor cells,36 and open new possible therapeutic scenarios based on this approach.10
Erythroid.
The presence of a split chimerism between mature erythrocytes and their progenitor has been recently described in persistent mixed chimera patients (M. Andreani, Haematologica, 2010, in press). In these patients, there was a predominant erythroid engraftment in the peripheral blood while the proportion of erythroid precursors (BFU-E [burst forming unit erythroid] colonies) and nucleated cells of donor origin in the bone marrow were equivalent. This observation of split chimerism between mature erythrocytes and their progenitors confirms that a limited HSC engraftment (up to 20%) bearing effective erythropoiesis is able to provide a sufficient amount of normal hemoglobin. Moreover, this limited engraftment with normal erythropoiesis is able to inhibit, through unknown mechanisms, the expansion of the ineffective thalassemic erythropoiesis, as well as the well-known associated clinical consequences.
Unfortunately, mixed chimera remains an extremely interesting and fascinating biological observation, which is not immediately applicable as a target for future medical treatments. However, it is a supporting observation, which may affect future developing gene therapy trial. It is a distinct possibility that, if researchers were able to transfer an efficient ß gene inside the thalassemic HSC, with this gene achieving a good level of expression, it would not be necessary to replace the entire diseased bone marrow to obtain clinical correction of the disease. In theory, with a 100% expression of the transfected-modified gene, a limited (20%) autologous gene-modified engraftment could be sufficient to obtain the required clinical effect.
Long-term Follow-up
Unfortunately, the hematologic cure of thalassemia by HSC transplantation fails to address thalassemia-related complications, which were already established at the time of transplantation (mainly iron overload and viral infection).
Iron overload and hepatitis C virus (HCV) infection are independent and mutually reinforcing risk factors for the progression of hepatic fibrosis and the development of cirrhosis. This was demonstrated by a prospective study that used yearly liver biopsy in transplanted thalassemic patients.40 Patients who were anti-HCV+ and also had high liver iron content had an 80% chance of developing progressive hepatic fibrosis within 10 years after successful transplantation. Conversely, patients with liver iron levels below 16 mg/g dry weight, who were also free of signs of HCV infection, showed no signs of progressive hepatic fibrosis.
The Problem of Relative Costs Between a Therapy Intended to Cure Versus a Lifelong Medical Therapy
Medical therapy of thalassemia is one of the most spectacular medical successes of the last three decades and has transformed thalassemia from a lethal disease of childhood to a chronic disease of adulthood, achieving at the same time prolonged survival with a good quality of life.46 In recent years, we have witnessed spectacular advances in our knowledge of iron overload pathophysiology and in our diagnostic capabilities. The development of effective and safe oral chelation regimen promises to further enhance these results.
However, the very large majority of patients live today in nonindustrialized countries, in which availability of these effective therapies is improving, but is still severely curtailed. A recent survey by Modell and Darlison47 indicated that more than 330,000 hemoglobinopathy-affected infants are born annually (83% sickle cell disorders, 17% thalassemias),47 and the large majority of these patients are born and live in the previously described areas of the world.
Availability of resources is a key issue for thalassemia treatment. A modern complete transfusion-chelation regimen is a high-technology therapeutic approach requiring unique expertise and resources.48 Medical therapy, without even considering the associated complications, is very expensive and not widely available. A recent Italian study, based on a cost/benefit ratio estimation from a societal perspective, quantified tariffs, expenses, and net earning in 2006 prices, and generated an estimated mean direct cost of medical therapy (transfusion + deferoxamine chelation) of approximately 15,000 euros per year per patient.49 In addition, proper medical therapy for thalassemia requires the support of an increasing number of advanced technologies (eg, cardiac magnetic resonance imaging) that have to be provided by an increasing number of treatment centers.48,50 HSC transplantation is also a high-skill/high-technology therapy, which is becoming increasingly available worldwide: the unpublished EBMT survey indicates that, presently, more than the half of the transplantation for thalassemia are performed outside industrialized/developed countries. Because the large majority of patients with severe thalassemia are now situated in either underdeveloped or still developing countries, the availability of resources is the main driver of therapeutic decisions. In this setting, transplant is likely to be the most cost-effective approach, but only for a limited number of patients.
Role of Transplantation in the Era of Oral Chelators
The central role of HSCT in thalassemia has been fully established through a long,51 controversial,52 and difficult, but scientifically productive, way. The animosities and controversies of the past should be reinterpreted as necessary and productive steps in a narrow and at times tortuous path, which eventually yielded outstanding clinical result and amazing improvement in the care and survival of our patients.
No prospective randomized clinical trials can be designed to provide a definitive answer to the challenge of choosing between transplant and medical therapy for each individual patient. In the absence of definitive evidence, the decision process is highly individualized and patient-specific. It must consider age, clinical status, willingness to undergo treatment, donor availability, capability and compliance to adhere the appropriate transfusion chelation regimen, quality of life, and resources. Parents of pediatric patients face an even more difficult decision (Fig. 4).

Factors that must be considered for individual decision making about HSCT for thalassemia.
Factors that must be considered for individual decision making about HSCT for thalassemia.
HSC transplantation is the only method available today to cure thalassemia major and other hemoglobinopathies. The development of oral iron chelators does not modify this position. However, much more uncertainty applies to the complex challenge of how the curative, but potentially lethal, HSCT treatment can be positioned as an alternative to a medical, noncurative therapy for adult and advanced disease patients. Transplantation outcomes today are much improved compared with the 1980s and 1990s, with more than 90% of patients surviving transplantation and more than 80% of them being disease-free in several centers worldwide.
Although long-promised gene therapy seems more and more imminent and feasible, we should remember all the lessons learned in the development of HSCT for thalassemia. If, as we all hope, gene therapy will provide, in the near future, the ultimate cure for thalassemia and other hematologic diseases, it will have done so by demonstrating at least its equipoise in terms of a cost/benefit ratio to HSCT, which remains today a widely applied, standard practice for the cure of thalassemia and other hemoglobinopathies.
Acknowledgment
The author thanks Carlo Brugnara, MD, for critical reading, revising, and making suggestions regarding this manuscript.
Disclosures
Conflict-of-interest disclosure: The author received an honoraria from and is a consultant for Novartis.
Off-label drugs: None disclosed.
Correspondence
Emanuele Angelucci, MD, Unità Operativa Ematologia e Centro Trapianti, Ospedale Oncologico di Riferimento Regionale “Armando Businco,” Via Edward Jenner, 09121 Cagliari, Italy; Phone: +390706092061; Fax: +390706095171; e-mail: emnang@tin.it