Abstract
Ultra high-risk myeloma can be defined as myeloma leading to death within 24 months. Despite tremendous improvements in the past decade (especially because of the availability of novel drugs such as thalidomide, bortezomib, and lenalidomide), these patients still represent 15% to 20% of the patients. Many prognostic factors can help to define these patients, including age, renal insufficiency, poor performance status, comorbities, International Staging System (ISS) stage 3, high proliferation, leukemic presentation, and acquired genetic changes, as defined by interphase fluorescence in situ hybridization or genomics. Several combinations of these prognostic parameters can define ultra high-risk patients, making a universal therapeutic proposal almost impossible. However, focusing on fit patients with ISS 3, high proliferation, and poor-risk genetic changes, these patients should probably benefit from dose-dense and prolonged therapeutic schemas, ideally within prospective trials.
For a long time, the prognosis of multiple myeloma (MM) has been poor with limited treatment options. Many studies reported a median overall survival of less than 3 years. The first glimpse of improved outcomes was reports in the 1990s of the use of high-dose melphalan with stem cell support.1–3 Using a polychemotherapy-based induction, followed by one or two courses of high-dose melphalan, the median overall survival improved to 5 years. However, more than one-half of the patients are not eligible for these intensive approaches. In the past decade, tremendous progresses have been observed, with the availability of three major new drugs: (1) thalidomide, (2) bortezomib, and (3) lenalidomide. With the use of these drugs, most commonly in combination with high-dose melphalan, patients now display a much better outcome.4–10 The latest estimates of median overall survival are probably around 10 years for patients under age 65 and around 5 to 6 years for older patients (Intergroupe Francophone du Myélome [IFM], personal communication, 2010). However, despite this remarkable progress, we have recognized that not all patients enjoy such a long survival. Some patients present with highly refractory MM or exhibit early relapses after an initial response. The three objectives of this paper are to (1) try and define who these patients are, (2) recognize these patients at the time of diagnosis, and (3) propose specific therapeutic approaches to improve their outcome.
Prognostic Parameters
The definition of these ultra high-risk patients is by definition arbitrary. This review focuses on patients with a median overall survival of less than 2 years. Many papers have described a huge number of prognostic factors in MM. Among this list, we can focus on those who have been confirmed by several studies. A nonexhaustive list comprises a high serum β2-microglobulin; a high C-reactive protein level; a low albumin level; a high creatinine level; a high lactate dehydrogenase level; a low hemoglobin level; a low platelet count; a high labeling index; leukemic presentation; poor cytogenetic parameters (eg, t(4;14), del(17p), or t(14;16)); and, as usual in oncology, older age. In this list, the most important parameters are probably β2-microglobulin,11–13 proliferation,14–16 and genetic abnormalities (Table 1).17–21 In a large cohort of more than 12,000 patients, the International Myeloma Working Group described an International Staging System (ISS) based on β2-microglobulin and albumin levels.13 This prognostic model classifies the patients into three groups, with different overall survival (62 months, 44 months, and 29 months for stages 1, 2, and 3, respectively). This system allows great progress in prognostication at the population level, but is more limited at the individual level. The weakness of this model is that it does not take into account intrinsic prognostic parameters, such as proliferation or genetic abnormalities. An ideal prognostic model would probably combine β2-microglobulin level (or ISS) that reflects tumor mass, renal insufficiency, and general patient condition, a marker of plasma cell proliferation (measured by labeling index or conventional cytogenetics) and genetic changes. The best way to identify these genetic changes is so far not defined. Many techniques can be used, including interphase fluorescence in situ hybridization (iFISH),17–21 gene expression profiling (GEP),22–24 or DNA copy number changes (analyzed by comparative genome hybridization [CGH] array or single nucleotide polymorphism [SNP] array).25,26 Most large series have used iFISH. The weakness of this technique in MM is that it requires plasma cell identification and that it allows the analysis of only a small number of chromosomal abnormalities. Despite these limitations, it has been shown that t(4;14) and del(17p) are major prognostic factors, at least in patients treated before the novel drug era.17–21 An updated ISS effort did show that karyotype abnormalities (reflecting high proliferation) and genetic changes (as identified by iFISH) significantly improved the ISS model (Enhancement of ISS Staging System incorporating genetic changes: an International Myeloma Working Group (IMWG) collaborative project, unpublished data). High-throughput molecular technologies (GEP and CGH array/SNP array) are probably more powerful, because they analyze all the abnormalities observed throughout the whole genome. However, these technologies require plasma cell purification and highly specifically dedicated platforms (including bioinformatics), and are not widely available to most physicians.
The next question is the stability of these prognostic factors in the novel drug era. Most (if not all) of them have been defined in retrospective cohorts of patients treated before the wide use of thalidomide, bortezomib, and lenalidomide. The current question is whether these prognostic parameters are still valid with novel treatment modalities?
Definition of Ultra High-Risk Patients
The ISS is probably still prognostic with novel combinations. Even though few studies specifically addressed this issue, most of the recently published trials showed that patients with ISS stage 3 displayed a shorter survival.7,10,27 This statement is probably also true for highly proliferative disease. Here, again, few studies did analyze this question. In patients treated at the University of Arkansas Total Therapy 3 program (polychemotherapy induction, double intensive melphalan, consolidation, and long-term maintenance with bortezomib-lenalidomide-dexamethasone cycles), an abnormal karyotype (indirectly reflecting plasma cell proliferation) is a powerful prognostic parameter.27 Similarly, a small study from the Mayo Clinic suggested that a high labeling index was still prognostic in patients treated with lenalidomide-dexamethasone.16
The same question addressed to genetic changes is still evolving. Some preliminary reports suggested that bortezomib was able to overcome the prognostic value of t(4;14). This has been first suggested in the VISTA (Velcade as Initial Standard Therapy in Multiple Myeloma) trial (comparing melphalan-prednisone vs bortezomib-melphalan-prednisone).28 In a very small number of patients (26 patients) presenting either t(4;14), del(17p), or t(14;16), time to progression and overall survival were similar in the bortezomib arm to that of patients lacking these chromosomal abnormalities (limiting the stability of the conclusions given limited power). The second report came from the Arkansas Total Therapy 3 program that showed that t(4;14), but also del(17p), were not anymore prognostic factors with this very peculiar therapy program.27,29 More recently, the IFM did show that a short-term bortezomib-dexamethasone (BD) induction significantly improved the outcome (both for event-free survival and overall survival) of patients with t(4;14), but not that of patients with del(17p).30 Nevertheless, even among bortezomib-treated patients, the t(4;14)(p16;q32) remains a significant prognostic factor Thus, based on iFISH and GEP analyses, t(4;14) is probably no longer a (strong) prognostic factor in patients treated with bortezomib. Because the VISTA trial and the Arkansas results showed a complete overcoming of the prognostic value of t(4;14), in contrast to the IFM data that showed only a partial improvement, it can be suggested that long-term treatment with bortezomib is recommended for these patients. Further validation, however, is needed. The case of patients with del(17p) is less clear. All of the recently published reports showed that del(17p) remains a strong prognostic factor.16,31 The only study suggesting that del(17p) was no longer prognostic came from the Arkansas group.29 Based on GEP analysis, using a surrogate proposal to detect -17p13, they showed that patients with a low TP53 expression encountered a similar outcome than patients with normal TP53 expression in the Total Therapy 3 program.
Some authors (especially those from Arkansas University) claim that all the patients should be analyzed by GEP to define a really strong prediction for survival. The first question is the feasibility of GEP in the routine practice. In a multicenter setting, only about half of the patients are analyzable with this technique (Avet-Loiseau H, Institut de Biologie, oral communication, September 17, 2010), because of the quality of the bone marrow samples sent to the laboratory. The second question is the definition of ultra high-risk patients using these technologies. Two reports on large cohorts of patients analyzed with GEP have been published so far (Figure 1).22,24 These two studies defined two different sets of genes identifying high-risk patients. Whether these models are identifying the same group of high-risk patients is an unresolved question. First, the two models were built on different platforms. Moreover, these models have been built on patients treated very differently. The Arkansas model was based on patients treated in the Total Therapy 2 program (polychemotherapy induction, double intensive melphalan, consolidation, long-term thalidomide maintenance), whereas the IFM model was built on patients treated with a vincristine-adriamycin-dexamethasone (VAD) induction, followed by double high-dose melphalan. A temptative cross-validation has been performed and showed a weak confirmation. This can be due to the different platforms, but also to the different therapeutic strategies. Despite these technical limitations, it seems that GEP is promising to define high-risk patients. Another study from the IFM using SNP array did show that some genomic changes (eg, 1q gains, trisomy 5, or 12p deletions) allowed identification of patients with very different outcomes.26 But, again, this technology requires plasma cell purification and is probably not usable for all the patients in a multicenter setting.

Genomic data showing the predictability of ultra high-risk myeloma patients using genomics. (A) Arkansas model using gene expression profiling. (B) IFM model using the SNP array.
Genomic data showing the predictability of ultra high-risk myeloma patients using genomics. (A) Arkansas model using gene expression profiling. (B) IFM model using the SNP array.
Ultra high-risk patients could be defined by ISS stage 3, a high plasma cell proliferation, and the presence of specific genetic changes, including del(17p) and probably other genomic abnormalities that need further work for a widely accepted definition. A specific statement is mandatory for the rare patients presenting with primary plasma cell leukemia. Even though no large study specifically dedicated for this patient population exists in the literature, most authors agree to consider these patients as high-risk (and probably ultra high-risk).32–34 Actually, these patients often combine a high β2-microglobulin level (or ISS stage 3), a high proliferative index reflected by the frequent abnormal karyotype and poor risk genetic abnormalities.
Management of Ultra High-Risk Patients
Even though the definition of ultra high-risk MMs varies from institution to institution, their management is very disappointing. For instance, in the Arkansas definition based on GEP, these patients represent 13% of all MMs. Even using extremely intensive and prolonged therapy, these patients display a very poor outcome.22 Using more “classical” therapies based on bortezomib-dexamethasone induction, followed by one or two courses of high-dose melphalan for younger patients, patients with del(17p) present a short, event-free survival and overall survival. Thus, how can this poor outcome be improved?
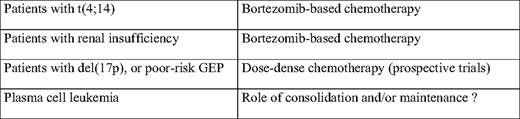
Several strategies have been tested. If considering the youngest patients (younger than age 65 or 70), several induction combinations (before high-dose therapy) have addressed this issue. These combinations included VAD, TD (thalidomide-dexamethasone), BD, or more complex strategies such as VTD-PACE (bortezomib-thalidomide-dexamethasone-cisplatinum-adriamycin-cyclophosphamide-etoposide). Despite some improvements in the complete response rates, a similar number of patients still died rapidly in all combination schemes. Then, some investigators worked on the high-dose regimen. A double-intensive strategy does not seem to benefit these patients.3 A combination of high-dose melphalan with bortezomib seemed to increase the response rate, but no data are available for overall survival.35 Finally, different consolidation/maintenance strategies have been tested. Thalidomide maintenance is definitely not the solution.36,37 Long-term maintenance with lenalidomide seems to improve the progression-free survival, but no data are available for overall survival (that could be shortened by the appearance of specific or overall resistances at relapse) or for high-risk patients (Attal M, personal communication, August 2010). Iterative long-term reinductions with bortezomib-lenalidomide-dexamethasone combinations appear promising for patients with t(4;14) or del(17p), but a population (representing about 13% of the patients) still escapes to this strategy and presents early death (Table 2).38
If a solution does exist, it will definitely be found through specific therapeutic trials. For instance, a trial testing dose-dense chemotherapy, prolonged over a long period of time, could be a solution to try to maintain a continuous therapeutic pressure on the clone. Another possibility could be to propose a frontline myeloablative stem cell transplant for such patients younger than age 50. These phase 2 trials should typically be multicentric, possibly international, to test different approaches over a short period of time. An important limit of this “trial” approach is that many of these poor-risk patients are not eligible for trial enrollment because of poor performance status, renal insufficiency, or comorbidities. Some investigators propose to test new drugs in these ultra high-risk patients. This proposal is highly debatable. The risk is to kill promising drugs tested on highly resistant patients. A strategy is recommended in which modern prognostic parameters (including genomics) would be analyzed in phase 2 and phase 3 trials testing novel drugs—eg, carfilzomib (second-generation proteasome inhibitor), pomalidomide (novel IMID [imidazoline]), heat shock protein, or histone deacetylase inhibitors—to specifically analyze their efficacy on high-risk patients.
Disclosures
Conflict-of-interest disclosure: The author is on the Board of Directors and Advisory Committees for Janssen-Cilag and Celgene.
Off-label drug use: None disclosed.
Correspondence
Hervé Avet-Loiseau MD, PhD, Professor, Laboratoire d'Hématologie, Institut de Biologie, 9 Quai Moncousu, 44093 Nantes Cedex 09, France; Phone: +33240087774; Fax: +33240084050; e-mail: herve.avetloiseau@chu-nantes.fr