Abstract
Essential thrombocythemia (ET) is a Philadelphia chromosome (Ph)–negative myeloproliferative neoplasm (MPN) characterized by thrombocytosis and megakaryocytic hyperplasia of the bone marrow, with presence of the JAK2 V617F mutation in 50%-60% of patients. ET evolves to myelofibrosis in a minority of cases, whereas transformation to acute leukemia is rare and increases in association with the use of certain therapies. Survival of ET patients does not substantially differ from that of the general population. However, important morbidity is derived from vascular complications, including thrombosis, microvascular disturbances, and bleeding. Because of this, treatment of ET must be aimed at preventing thrombosis and bleeding without increasing the risk of transformation of the disease. Patients are considered at high risk of thrombosis if they are older than 60 years or have a previous history of thrombosis and at high risk of bleeding if platelet counts are > 1500 × 109/L. Patients with low-risk ET are usually managed with low-dose aspirin, whereas treatment of high-risk ET is based on the use of cytoreductive therapy, with hydroxyurea as the drug of choice and IFN-α being reserved for young patients or pregnant women. For patients resistant or intolerant to hydroxyurea, anagrelide is recommended as second-line therapy. Strict control of coexistent cardiovascular risk factors is mandatory for all patients. The role in ET therapy of new drugs such as pegylated IFN or the JAK2 inhibitors is currently under investigation.
Introduction
Essential thrombocythemia (ET), first recognized as a distinct entity in 1934 under the term “hemorrhagic thrombocythemia,” is a myeloproliferative neoplasm (MPN) characterized by thrombocytosis with bone marrow megakaryocytic hyperplasia and a tendency to develop vascular complications, including thrombosis, microvascular disturbances, and hemorrhage. ET was placed by Dameshek in 1951 within the myeloproliferative disorders, together with chronic myeloid leukemia, polycythemia vera (PV), and primary myelofibrosis (PMF).1 The discovery of the Philadelphia (Ph) chromosome led to coining of the term “classic” Ph-negative myeloproliferative disorders to encompass ET, PV, and PMF, three heterogeneous disorders with clonal origin in a multipotent hemopoietic stem cell that share different clinical, hematologic, and biological features. Transition from one disorder to the other is sometimes observed. In 2005, the discovery of an acquired mutation in the JAK2 gene in a high proportion of patients with classic Ph-negative MPNs2–5 provided a molecular basis for Dameshek's intuitive integration of these entities. ET is the most common of the three disorders and the one with a more favorable prognosis, because it affects the patients' quality of life more than their survival due to the increased occurrence of vascular complications.6 Evolution to myelofibrosis is observed in 4%-8% of patients at 10 years,7,8 whereas leukemic transformation is rare but can increase with the use of certain cytoreductive drugs.9,10 Therefore, ET treatment should be aimed at preventing thrombosis and bleeding but without increasing the risk of transformation. This article summarizes the current status of ET therapy and emphasizes the importance of a risk-adapted treatment strategy in the management of these patients.
Pathogenesis
A major milestone for the understanding of MPN pathogenesis was the identification of the JAK2 V617F mutation in the majority of PV patients and in 50%-60% of those with ET and PMF.2–5 This mutation produces an increased tyrosine kinase activity of JAK2, resulting in a cytokine-independent phenotype. Although the JAK2 mutation is critical for MPN appearance, molecular analysis has indicated that it may often arise as a secondary event after the acquisition of other mutations (reviewed in Vannucchi and Guglielmelli11 ). It is also intriguing why the same molecular lesion produces three different phenotypes. According to the most widely accepted theory, the “gene-dosage hypothesis,” the mutated allele burden is the more important contributor to MPN phenotype.11 This theory stems from the observation that the higher JAK2 mutated allele burden is found in PV and PMF and the lower in ET, whereas homozygosity for the JAK2 mutation is frequent in PV and PMF and rare in ET. Moreover, experimental studies in animal models parallel the findings in human MPNs. Therefore, overexpression of JAK2 V617F in mice results in a picture mimicking PV without thrombocytosis, with further evolution to myelofibrosis, whereas low JAK2 V617F expression produces an ET-like disease with thrombocytosis but not erythrocytosis.12 However, the allele burden is not sufficient to explain the MPN phenotype. It is quite likely that other factors, including pre-JAK2 mutation events, host genetic factors such as certain polymorphisms, epigenetic factors, additional somatic mutations not yet identified, and host modifiers such as iron availability, contribute to the final MPN phenotype.11
Whereas the JAK2 V617F mutation is the molecular abnormality more frequently found in ET, 3%-5% of patients display mutations in the thrombopoietin receptor gene or the MPL gene (from the myeloproliferative leukemia virus oncogene, which induces a PMF-like with thrombocytosis in mice).13,14 These mutations (MPL W515L/K) are associated with a gain of function and are also found in approximately 5%-10% of PMF patients, but not in PV. Experiments in mice transplanted with hemopoietic precursors harboring the MPL mutation show that they develop a rapidly evolving disease with leukocytosis, thrombocytosis, splenomegaly, and BM fibrosis, but not erythrocytosis.
Familial occurrence of MPNs has long been known. Hereditary predisposition was supported by an epidemiological study from Sweden showing an increased occurrence of these diseases among relatives of MPN patients.15 Recently, molecular support for these clinical observations was provided by the discovery of an increased frequency of a special haplotype of the JAK2 gene, the 46/1 haplotype, in MPN patients and their first-degree relatives.16
Diagnosis and clinical findings
The MPN diagnostic criteria were updated in 2007 by an ad hoc committee on behalf of the World Health Organization (WHO) to reflect the new molecular developments in these diseases.17 In the case of ET, the diagnostic platelet count threshold was also reduced from 600-450 × 109/L. Table 1 shows the current WHO diagnostic criteria for ET, and it can be seen that the diagnosis of ET remains one of exclusion. Therefore, in addition to thrombocytosis and megakaryocytic hyperplasia of the BM, the remaining criteria are destined to rule out other MPNs, some myelodysplastic syndromes, and, if molecular alterations are lacking, iron deficiency or causes of reactive thrombocytosis. When iron deficiency is found in a subject with isolated thrombocytosis, iron supplementation is mandatory to exclude iron deficiency–driven thrombocytosis (if no molecular alteration is found) or underlying PV with normal erythroid values if the JAK2 mutation is present.
The differential diagnosis between ET and the so-called “pre-fibrotic” form of PMF17 is controversial. This latter condition, which presents with thrombocytosis, but without anemia, splenomegaly, leukoerythroblastic blood, or BM fibrosis, is characterized by the presence of dysplastic megakaryocytes similar to those in overt PMF. The recognition of such an entity has been criticized18 because its diagnosis is subjective, involving issues of reproducibility, whereas its long-term implications are not well known. In a recent collaborative study in which the clinical, laboratory, and histologic features of 1104 ET patients were retrospectively reviewed, 16% were reclassified as having “pre-fibrotic” PMF and these patients more frequently evolved to myelofibrosis or leukemia and had shorter survival.19 It is likely that a minority of ET patients could have this histological entity, but for now, their management should be guided by their clinical findings and evolution rather than by the histological features. Therefore, unless these patients do not develop clinically overt myelofibrosis, they should be managed as ET patients.
ET is the most frequent of the classic MPNs, a fact related to the widespread use of routine laboratory studies and further accentuated by the recent reduction in the platelet count diagnostic threshold.20 The disease appears at all ages, with a median age of ∼60 years, and shows a female predominance. Most patients are asymptomatic at presentation and can remain so for years. Vascular events, especially thrombosis and microvascular occlusive symptoms, are the more frequent clinical manifestations8,21,22 ; however, their incidence is difficult to establish given the heterogeneity in methodology, follow-up, and therapy in published studies. Thrombosis is more often arterial than venous. Bleeding is usually associated with thrombocytosis exceeding 1500 × 109/L due to acquired von Willebrand disease.23 Aquagenic pruritus is occasionally seen, generally in JAK2-positive patients.
The molecular heterogeneity of ET involves certain phenotypic heterogeneity.24,25 Therefore, patients with the JAK2 mutation display higher hemoglobin levels and neutrophil counts and lower platelet counts and serum erythropoietin levels, a phenotype resembling PV, and are more prone to develop vein thrombosis and to evolve to PV. Moreover, among JAK2-positive patients, those with higher transcripts more frequently have pruritus, splenomegaly, early need for cytoreduction, and a higher tendency to myelofibrosis. Patients with MPL mutations have lower hemoglobin (W515L) or higher platelets (W515K) and a higher frequency of microvascular disturbances.26
Pathogenesis of thrombosis
The pathogenesis of the thrombosis seen in ET is not fully understood. Table 2 summarizes the factors that have been implicated in the appearance of this complication. Advanced age and previous history of thrombosis are the more important determinants of thrombosis.8,20,21 Other host factors, such as coexistent vascular risk factors (arterial hypertension, diabetes, dyslipidemia, and smoking) and occasional thrombophilia may also contribute to the thrombotic risk. No correlation has been found between platelet values and the risk of thrombosis. In recent years, the possible importance in MPN thrombosis of leukocytes and JAK2 mutational status and allele burden has been a field of intense investigation (reviewed in Cervantes et al6 ). In brief, although the results of most such studies tend to support a contributory role of higher leukocyte counts and JAK2 mutation status and allele burden in thrombosis, agreement is not universal. The role of leukocyte and platelet activation is better established (reviewed in Cervantes et al27 ). Indeed, leukocytes of ET patients have an activated phenotype, with increased phagocytosis, overexpression of the membrane CD11b antigen and leukocyte alkaline phosphatase, and increased plasmatic and cellular elastase content. Overexpression of monocyte tissue factor, currently considered as the main trigger of the coagulation cascade in vivo, has been demonstrated in ET patients with thrombosis, and an increased number of platelets expressing P-selectin has also been found. The link between leukocyte and platelet activation and thrombosis in ET has been reinforced by the finding of increased concentrations of markers of coagulation and endothelial activation in patients with increased blood cell activation.28,29 These findings support the notion that activation of the leukocytes (notably, the monocytes) and, to a lesser extent, the platelets may be involved in the genesis of the thrombosis of ET. In addition, a possible association between the latter and the JAK2 mutation or its allele burden is increasingly emerging.
Prognostic factors and risk stratification
Survival of ET patients does not seem to differ substantially from that of the general population,6,30 although a certain reduction after the first decade has been reported.31 Evolution to myelofibrosis is seen in 3.9%-8.3% of patients at 10 years7,8 and in more than 15% at 15 years.7 In one study, the use of anagrelide was associated with an increased frequency of evolution to myelofibrosis.32 Transition to PV is occasionally observed. Acute myeloid leukemia and myelodysplasia can occur in treatment-naive patients, but they are more frequently seen in those having received radioactive phosphorus or pipobroman.9,10
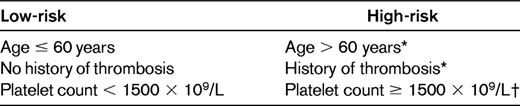
Vascular events represent by far the most frequent complication of ET, but they affect the patients' quality of life more than their survival. Because of this, prognostic stratification of ET must be dictated by the patients' risk of developing such complications, and therefore the treatment goal should be to prevent thrombosis and bleeding without increasing the risk of transformation of the disease.33 In accordance with the recent recommendations from the European Leukemia Net expert panel,33 Table 3 shows the most widely accepted prognostic stratification of ET. As can be seen, on the basis of the patients' risk of thrombosis or bleeding, a high- and a low-risk group are recognized. It must be pointed out that attempts to consider an intermediate-risk group, which would include patients from 40-60 years of age with no high-risk factors and patients younger than 60 years with cardiovascular risk factors, have not gained general acceptance. Whether these patients should receive cytolytic therapy remains a matter of controversy. The results of an ongoing PT-1 trial comparing hydroxyurea plus low-dose aspirin with low-dose aspirin alone in this ET subpopulation will hopefully provide evidence to help answer this question.
Therapy
Therapeutic options for ET patients range from the most conservative approach (careful observation) to low-dose aspirin and, in the upper extreme, cytoreductive drugs. Whereas the evidence supporting some of the available options is limited, there is general consensus on the use of a risk-adapted strategy in the treatment of these patients, restricting cytoreductive therapy to patients with high risk of thrombosis or bleeding. Figure 1 shows a therapeutic algorithm for ET based on this strategy. Irrespective of the individual thrombohemorrhagic risk, strict control of generic cardiovascular risk factors, including smoking cessation, is mandatory in all patients. Treatment is also required for symptoms due to microvascular occlusion, including erythromelalgia, acroparesthesia, and digital ischemia; neurological symptoms such as transient ischemic attack, migraine-like headache, and dizziness; and vision disturbances such as transient blindness, blurred vision, or photopsia. In addition, specific approaches are required for special situations such as pregnancy, pediatric ET, surgery, or splanchnic vein thrombosis.

Management of low-risk patients
The demonstration in the European Collaboration on Low-dose Aspirin in Polycythemia Vera study34 that the risk of thrombosis was reduced by antiplatelet therapy with low-dose aspirin, as well as the fact that combination of the latter with hydroxyurea was established as the choice treatment for high-risk ET by the results of the PT-1 study,35 fostered the widespread use of low-dose aspirin as primary prophylaxis of thrombosis in low-risk ET. However, the evidence for the role of such a strategy in ET is weak. Actually, in some studies, the incidence of thrombosis in untreated patients did not differ from that observed in a healthy control population.36 A recent study has provided some guidance for indications for low-dose aspirin in low-risk ET.37 The investigators retrospectively analyzed the results of antiplatelet therapy (mostly low-dose aspirin) or careful observation in 300 low-risk ET patients with a follow-up of 802 and 848 person-years, respectively, showing that antiplatelet therapy reduced the risk of venous thrombosis in JAK2-positive patients and the risk of arterial thrombosis in those with cardiovascular risk factors, whereas it was associated with an excess of bleeding episodes in patients with platelet counts above 1000 × 109/L. The retrospective nature of the study and the fact that antiplatelet therapy was based on the physician's best judgement pose some limitations to the results of the above study. However, the high number of patients analyzed and the long follow-up make its conclusions of value. Therefore, although the simplest approach could be to administer low-dose aspirin to all low-risk ET patients in the absence of a major contraindication, a reasonable strategy could be to give low-dose aspirin to low-risk patients except those with contraindication for aspirin or with platelet counts more than 1000 × 109/L to minimize the risk of hemorrhage.
In addition to its role in the prophylaxis of thrombosis, low-dose aspirin is the treatment of choice for the microvascular disturbances of ET, being especially effective in erythromelalgia and transient neurological manifestations.38 The available evidence points to the superiority in this setting of low-dose aspirin more than new antiplatelet agents such as clopidogrel or ticlopidine.39 If no effective control of the microvascular symptoms is achieved with aspirin, then the use of platelet-lowering agents can be considered.
Management of high-risk patients
As mentioned previously, there is general agreement on the administration of cytoreductive therapy to all patients with high-risk ET. Hydroxyurea became the first-line drug of choice after a randomized trial showed that it produced a significant reduction in thrombotic events.40 In that study, 114 high-risk patients were randomly assigned to receive hydroxyurea or no cytoreductive therapy and, after a median follow-up of 27 months, thrombosis rate was 3.6% in treated patients versus 24% for untreated patients. This observation was further supported by the results of the PT-1 trial, a randomized study that compared hydroxyurea plus aspirin versus anagrelide plus aspirin in 809 patients with high-risk ET.35 After a median follow-up of 39 months, the results demonstrated the superiority of hydroxyurea plus low-dose aspirin in preventing vascular events overall and transformation to myelofibrosis, although anagrelide proved to be superior in preventing venous thrombosis. Because anagrelide was equally effective in reducing the platelet counts, the superiority of hydroxyurea might be ascribed to its ability to suppress not only the platelets but also the leukocytes. Conversely, although there have been concerns about the possible mutagenic effect of hydroxyurea, such an effect has not been convincingly proven. Actually, many of the cases of transformation to acute leukemia or myelodysplasia occurred in patients who had also received other drugs, whereas biological studies have failed to demonstrate an increase in DNA mutations in MPN patients receiving hydroxyurea. However, it seems prudent to avoid hydroxyurea as much as possible in patients under the age of 40 years, given the remaining uncertainties about its leukemogenic potential in the long term. The starting hydroxyurea dose is 500 mg twice daily, with further adjustment to maintain normal platelet counts (without provoking clinically relevant neutropenia or anemia) and to eliminate the signs and symptoms of the disease. Strict criteria of response were defined by a panel of experts from the European Leukemia Net for their application in clinical trials.41 However, it is uncertain whether achievement of such stringent criteria translates into superior patient benefit in clinical practice.22,42 At the usual dose, hydroxyurea is generally well tolerated, but intolerance can develop, including anemia, leukopenia, and the appearance of oral or leg ulcers, skin lesions, or gastrointestinal symptoms.
For high-risk ET patients resistant or intolerant to hydroxyurea, anagrelide, an imidazoquinazoline derivative, is considered as the second-line therapy. The drug selectively reduces the platelet counts by inhibiting megakaryocytic differentiation and has also some effect on platelet aggregation. The latter effect is the most likely explanation for the higher rate of hemorrhage observed in the anagrelide plus aspirin arm of the PT-1 study and would preclude the combination of anagrelide with aspirin. Although the preliminary results of the ANAHYDRET study,43 which prospectively compared hydroxyurea and anagrelide in ET, failed to show inferiority of anagrelide to hydroxyurea, it must be noted that the number of patients in that study was small and the follow-up relatively short. The starting anagrelide dose is 0.5 mg given 2 or 3 times daily, with progressive increase based on platelet counts and side effects. The latter led to treatment discontinuation in up to one-third of patients and included headache, palpitations, arrhythmia, fluid retention, heart failure, and anemia. Caution for anagrelide use is required in elderly patients and in those with cardiac disease.
Other drugs occasionally used in ET are radioactive phosphorus, pipobroman, and busulfan. Given their leukemogenic potential (clearly demonstrated for the first two drugs),9,10 they should be restricted to patients with reduced life expectancy, usually those over the age of 75 years who cannot tolerate hydroxyurea. Recombinant IFN-α can control platelet counts but is associated with frequent side effects, which makes its long-term administration difficult, especially as the patient's age increases. Because of this, and given its lack of leukemogenic and teratogenic potential, IFN should be reserved for young patients with high-risk ET and for women wishing to become pregnant or with high-risk pregnancy.
Management of special situations
ET is associated with an increased risk of miscarriage and serious complications of pregnancy, such as pre-eclampsia, placental abruption, intrauterine death or stillbirth, and intrauterine growth retardation; venous thrombosis may also occur, especially in the postpartum period.44 Based on the presence or not of a history of thrombosis, ET-related bleeding or serious problems in previous pregnancies, high- and low-risk pregnancies are distinguished in women with ET.33 Low-risk pregnancy can be managed with low-dose aspirin throughout the pregnancy (unless contraindicated) and low-molecular-weight heparin (LMWH) during the 6 weeks after delivery. Management of high-risk pregnancy must be more aggressive, including anticoagulation with LMWH during the pregnancy and for 6 weeks after delivery, as well as cytoreduction with IFN in case of previous major thrombosis, recurrent miscarriage, serious obstetric problems, or marked thrombocytosis.
ET is not infrequent in children. Although the diagnostic criteria are the same as in adults, in cases of familial occurrence, screening for rare mutations is recommended to differentiate JAK2-negative ET from rare familial disorders due to mutations of thrombopoietin or MPL other than W515. Because the majority of children with ET have low-risk disease, cytoreduction is rarely indicated. It is important to take into account that aspirin must be avoided as much as possible in children below the age of 12 years because of the risk of provoking Reye syndrome. In addition, IFN can be especially dangerous in this population because of its side effects, including flu-like syndrome, neuropsychiatric symptoms, or the possible development of autoimmune disorders. Moreover, although no case of transformation has been reported in MPN children treated with hydroxyurea, the possible leukemogenicity of this drug in the long term is also of concern. Because of this, in children with ET, cytoreductive therapy should be used only as a last resort.33
In ET patients who must undergo surgery, antiplatelet therapy must be stopped 7-10 days before the procedure. In case of elective surgery, normalization of the platelet counts by cytoreductive therapy is also recommended. For all patients, prophylaxis of postoperative thrombosis with LMWH is mandatory.33
Treatment of splanchnic vein thrombosis should include long-life anticoagulation and administration of hydroxyurea to keep the platelet counts below 400 × 109/L.33
Investigational drugs
In an attempt to minimize IFN toxicity, a study from the M. D. Anderson group provided the pegylated isoform of IFN-α2a to 39 ET patients who had previously received at least one cytoreductive drug.45 Hematologic response was 92%, including 86% complete responses, and some molecular responses were also seen. Tolerability was better than that of standard IFN, because only 10% of patients dropped out of the study due to IFN-related side effects. A retrospective study from the French Intergroup on Myeloproliferative Disorders that included 59 patients with high-risk ET treated for a median of 16 months reported 92% hematologic responses, which were complete in 76% of cases, with 81% of patients remaining on treatment at last follow-up.46 However, toxicity of pegylated IFN is not negligible and its effects in the long run are not yet known. Hopefully, the results of an ongoing international phase 3 study comparing pegylated IFN-α2a with hydroxyurea in newly diagnosed patients with high-risk ET will allow the establishment of the role of pegylated IFN-α2a in these patients.
The discovery of the JAK2 mutation triggered the development of a molecularly targeted therapy for the MPNs with the hope of reproducing the success of the tyrosine kinase inhibitors in chronic myeloid leukemia. For now, the experience with the use of JAK2 inhibitors in ET patients who are not in the myelofibrotic phase is limited. Ruxolitinib (formerly known as INCB018424)47 achieved normalization of platelets counts in 49% of 39 ET patients resistant or intolerant to hydroxyurea after a median of 0.5 months, with 82% of them maintaining the platelets below 600 × 109/L after a median follow-up of 15 months. A higher than 50% JAK2 allele burden decrease was seen in 12% of patients. Clinical responses were unrelated to the presence or absence of the JAK2 mutation or changes in its allele burden.
Another JAK2 inhibitor, CEP701, showed lower efficacy than ruxolitinib, whereas it was associated with substantial gastrointestinal toxicity, mainly consisting of diarrhea and nausea.48 Studies with newer JAK2 inhibitors are at an early stage.
Finally, the role of novel drugs in high-risk ET, such as the histone-deacetylase inhibitors givinostat and vorinostat and the telomerase inhibitor imetelstat, is currently being investigated.
Future directions
Unlike in other MPNs (notably PMF), the medical needs of ET are mostly covered, with several drugs being available for patients requiring cytoreduction. It is therefore unlikely that the treatment of ET will change substantially over the coming years. This would occur only if some of the newer drugs under investigation show a high efficacy in the achievement of molecular responses in other JAK2-positive MPNs, and if this translates to a lower incidence of vascular complications. Given the good prognosis of ET, prolonged follow-up will be required to demonstrate such an effect in this disease.
Acknowledgments
The author acknowledges the contribution to this field of many researchers whose work has not been quoted due to space restrictions, as well as the support from grants Retics RD06/0020/004 and FIS PI10/00236 from the Instituto de Salud Carlos III, Spanish Ministry of Health.
Disclosures
Conflict-of-interest disclosure: The author is a consultant for Novartis, Bristol-Myers-Squibb, Pfizer, Celgene, MSD, and Astra-Zeneca, and is affiliated with the speakers' bureaus for Novartis, Bristol-Myers-Squibb, and Shire. Off-label drug use: Experimental use of JAK2 inhibitors and IFN in ET treatment.
Correspondence
Francisco Cervantes, MD, PhD, Hematology Department, Hospital Clínic, Villarroel 170, 08036 Barcelona, Spain; Phone: 34-932275428; Fax: 34-932275484; e-mail: fcervan@clinic.ub.es.