Abstract
Despite recent attempts to define and classify patients with marked eosinophilia and features consistent with myeloproliferative disease, areas of controversy remain. These are particularly apparent in situations in which multiple lineages are involved in a clonal process and clinical manifestations are overlapping. Although the introduction of new molecular diagnostics and targeted therapies has begun to clarify the boundaries between some of these disorders, several questions remain with respect to the classification of patients with myeloproliferative hypereosinophilic syndrome (HES) of unknown etiology.
Introduction: history
Hypereosinophilic syndrome (HES) was first recognized as a distinct clinical entity in 1975, when Chusid et al1 proposed the following definition: (1) a persistent eosinophilia of 1.5 × 109/L eosinophils/mm3 for longer than 6 months or death before 6 months associated with the signs and symptoms of hypereosinophilic disease; (2) a lack of evidence for parasitic, allergic, or other known causes of eosinophilia; and (3) presumptive signs and symptoms of organ involvement, including hepatosplenomegaly, organic heart murmur, congestive heart failure, diffuse or focal nervous system abnormalities, pulmonary fibrosis, fever, weight loss, and anemia. Even then, it was recognized that these criteria encompassed a wide variety of disorders, with chronic eosinophilic leukemia (CEL) at one end of the spectrum. In fact, nearly 1/3 of the cases described in this landmark paper had detectable myeloblasts in the peripheral blood, and 4 of 18 subjects had abnormal cytogenetics.
More recently, the availability of molecular and immunologic diagnostic techniques and targeted therapies has led to the identification of specific etiologies in some patients with HES. This has caused considerable controversy, particularly with respect to the subset of eosinophilic patients with evidence of chronic myeloproliferative disease. Whereas the 2001 World Health Organization (WHO)2 classification lumped all patients with CEL or HES into a single diagnosis under the category of chronic myeloproliferative diseases, the new WHO classification separates these patients into 2 groups: CEL not otherwise categorized (CEL-NOS) and myeloid neoplasms associated with eosinophilia and abnormalities of PDGFRA, PDGFRB, and FGFR12 (Table 1). CEL-NOS is listed in the category of myeloproliferative neoplasms, whereas the second group is a new category. Patients with HES and myeloproliferative features who do not have detectable mutations or evidence of clonality are not included in the 2008 classification scheme. Although this classification makes sense in some respects, including the recognition that D816VKIT-positive systemic mastocytosis (SM) is a disorder that is distinct from PDGFRA-associated myeloid neoplasms, it is confusing from a clinical diagnostic and treatment standpoint because there is considerable overlap between the clinical and hematologic manifestations of CEL-NOS, PDGFRA-associated disease, and those of patients with HES who have myeloproliferative features but in whom clonality cannot be proven.
An alternative classification scheme, proposed by the Hypereosinophilic Diseases Working Group of the International Eosinophil Society in 20064 and revised at a subsequent workshop in 2010,5 attempted to incorporate both clinical and molecular features (Table 1). Patients presenting with HES with clinical, laboratory, and BM features consistent with a myeloproliferative disorder were grouped under the heading “myeloproliferative forms.” This category was divided into CEL and myeloproliferative HES based on the presence of eosinophil clonality as demonstrated by molecular tests, cytogenetics, HUMARA assay, or the presence of increased blasts. The purpose of this classification scheme was 2-fold: (1) to emphasize that patients with myeloproliferative features with or without demonstrable eosinophil clonality are more similar to each other than to other patients with HES and (2) to minimize the need for reclassification of patients as new molecular abnormalities are defined. Myeloid neoplasms, including those associated with FGFR1 and KIT, were included in a separate category called “associated” to indicate that marked eosinophilia can be present, but that the predominant clinical features are due to involvement of lineages other than eosinophils.
Regardless of the classification system used, it is clear that these disorders are rare. In fact, a recent study using the Surveillance, Epidemiology and End Results (SEER) database calculated the crude incidence of HES including CEL (under the general category of chronic myeloproliferative disorders) at 0.036 in 100 000 person-years.6 This has further complicated efforts to draw general conclusions about clinical manifestations, responses to treatment, and prognosis in affected patients.
This review describes 3 distinct WHO-defined disorders for which the clinical and/or hematologic presentations overlap with HES and discuss the relationship of these disorders to “myeloproliferative HES.” Myeloid neoplasms with abnormalities of PDFRB, FGFR1, and JAK2 will not be discussed herein, because these disorders rarely present as HES despite the presence of peripheral and/or BM eosinophilia.
Myeloproliferative HES
A subgroup of patients with eosinophilia > 1.5 × 109/L, features of myeloproliferative disease, and poor prognosis has long been recognized.7 In fact, almost 50% (33 of 72) of patients who met Chusid's criteria for HES in 2 separate series reported in the early 1980s were found to exhibit features of a myeloproliferative disorder, including hypercellularity of the BM, abnormalities in cell lineages other than eosinophils, myelofibrosis, splenomegaly, and elevated serum B12 levels.8,9 These patients were less likely to respond to steroid therapy, had more aggressive disease, and included 8 patients with detectable cytogenetic abnormalities (CEL-NOS). More recently, myeloproliferative features in the setting of marked eosinophilia and clinical manifestations of HES have been associated with the presence of the FIP1L1/PDGFRA fusion gene.10
Although it is clear that CEL-NOS- and PDGFRA-associated disease account for a significant proportion of the cases of HES with myeloproliferative features, a causative genetic abnormality cannot be demonstrated in all cases. In a French series of 35 patients with HES, 9 patients were described as having clinical or hematological features of myeloproliferative syndrome (including palpable splenomegaly, neutrophilia, circulating myelocytes and/or erythroblasts, hyperplastic BM, and myelofibrosis).11 Three of the 9 patients had no detectable FIP1L1/PDGFRA (F/P) fusion gene or cytogenetic abnormalities, and 1 responded to imatinib therapy. Several additional case series have described imatinib-responsive, F/P-negative patients with HES,12–15 although the presence of myeloproliferative features in these patients has not been systematically addressed.
Therefore, despite early data suggesting that HES with myeloproliferative features has an aggressive course and poor prognosis, it is unclear whether this is the case when patients with cytogenetic abnormalities and mutations in PDGFRA are excluded. Whether these patients should be classified separately from other patients with “idiopathic HES” and/or managed differently remains controversial.
PDGFRA-associated disease
Myeloid neoplasms associated with eosinophilia and abnormalities of PDGFRA account for approximately 10%-20% of patients presenting with clinical findings consistent with HES. First described in a patient with imatinib-responsive HES and a t(1;4)(q44;q12) translocation,13 the F/P fusion gene is the most common abnormality and arises from an 800-kb interstitial deletion, del(4)(q12q12), that leads to constitutive activation of PDGFRA. The break points in FIP1L1 are variable, but are typically located in a 40-kb region spanning introns 7-10 of FIP1L1. In contrast, the break points in PDGFRA appear to be restricted to a region of exon 12 that contains the WW-like region of the juxtamembrane domain. Although some patients with F/P have a reciprocal translocation in 4q12, most have a normal karyotype. More recently, several additional PDGFRA fusion partners have been identified, including KIF5B,16 CDK5RAP2,17 STRN,18 BCR, and ETV6.18 Although point mutations in PDGFRA have been detected in patients with HES and have been shown to cause growth factor–independent proliferation in vitro and leukemia-like disease in mice, their role in disease pathogenesis in humans is unproven.19
Clinical and laboratory features
The overwhelming majority of patients with PDGFRA-associated myeloid neoplasms are male, although the reason for this gender bias is unknown. Whereas most patients are between 20 and 40 years of age, the F/P fusion has been described in children as young as 3 months20 and in the elderly. End-organ manifestations are similar to those seen in classic HES, although splenomegaly and fibrotic complications, including endomyocardial fibrosis, restrictive pulmonary disease, and myelofibrosis, seem to be more frequent in patients with the F/P mutation.10–12,21 Unusual skin manifestations, including lymphomatoid papulosis22,23 and mucosal ulcerations,10,24 have also been reported. Before the availability of imatinib, prognosis for patients with PDGFRA-associated eosinophilic disorders was poor, with a 30%-50% mortality at 5 years, primarily due to cardiac and neurologic complications.10,21
As in other myeloid neoplasms, multiple lineages can be involved in the clonal process.25 Despite this, proliferation is generally restricted to eosinophils, mast cells, and in approximately 50% of cases, neutrophils. T-cell clones,26,27 as well as synchronous T-cell lymphoblastic lymphoma,28 have been described in patients with F/P-positive myeloid neoplasms, although this appears to be a rare phenomenon. Laboratory abnormalities other than leukocytosis that are commonly seen in PDGFRA-associated myeloid neoplasms include anemia, thrombocytopenia, and elevated serum B12 and tryptase levels.10,11,21 Serum IgE elevation is variable. BM examination typically reveals a hypercellular BM with marked eosinophilia and increased numbers of scattered, spindle-shaped, CD25+ mast cells without dense focal infiltrates29 (Figure 1). Reticulin fibrosis is often increased.

BM histopathology in D816V KIT-positive SM with eosinophilia (D816V KIT) and PDGFRA-positive myeloid neoplasia (FIP1L1-PDGFRA). Sections were stained with H&E or anti-tryptase antibody. Dense aggregates containing > 15 mast cells are seen in D816V KIT-positive SM with eosinophilia, whereas scattered spindle-shaped mast cells forming loose collections are typical of PDGFRA-positive myeloid neoplasia. Magnification is 100×.
BM histopathology in D816V KIT-positive SM with eosinophilia (D816V KIT) and PDGFRA-positive myeloid neoplasia (FIP1L1-PDGFRA). Sections were stained with H&E or anti-tryptase antibody. Dense aggregates containing > 15 mast cells are seen in D816V KIT-positive SM with eosinophilia, whereas scattered spindle-shaped mast cells forming loose collections are typical of PDGFRA-positive myeloid neoplasia. Magnification is 100×.
Diagnosis
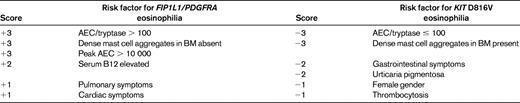
The F/P mutation can be detected by RT-PCR or FISH.13 Although a formal comparison study has not been performed to date, there does not seem to be a difference in sensitivity or specificity between the 2 methods, and the fusion can be detected equally well in peripheral blood and BM aspirates. Other PDGFRA fusions described to date have been associated with translocations and can be detected with routine cytogenetic analysis. Despite overlap between the diagnostic criteria for SM (see below) and the characteristic features of PDGFRA-associated myeloid neoplasms, it is important to distinguish between these 2 disorders because there are differences in clinical manifestations, response to therapy, and prognosis. Although demonstration of a D816V mutation in KIT or a PDGFRA fusion gene is clearly the “gold standard” for diagnosis, SM and PDGFRA-associated myeloid neoplasms can also be differentiated using a clinical scoring system (Table 2).29
Treatment and monitoring
Imatinib is the treatment of choice for patients with PDGFRA-positive disease, and the majority of patients achieve clinical and hematologic improvement within 2-4 weeks and molecular remission within 3-6 months. Although most clinical manifestations improve with imatinib treatment, structural damage, including cardiac valve abnormalities and ischemic injury, are permanent. Therefore, imatinib treatment should be initiated as soon as possible after the diagnosis is made.
Although there is some controversy as to the most appropriate starting dose (100-400 mg daily) of imatinib, it is clear that most patients can be maintained successfully on low doses (100 mg daily to 100 mg weekly) once remission is achieved.30 Side effects of therapy are similar to those seen in the treatment of chronic myeloid leukemia (CML), with the exception of acute cardiac decompensation, which has been described after imatinib treatment of HES in patients with known cardiac involvement and/or an elevated serum troponin.31 Administration of corticosteroids for several days before the initiation of imatinib therapy and for several days thereafter has been recommended, although there are no data to confirm that this will prevent complications in patients with preexisting eosinophilic infiltration of the heart, and myocardial necrosis has occurred after initiation of imatinib therapy in at least one patient despite high-dose steroids.32 Imatinib-induced cardiac toxicity independent of eosinophilia was demonstrated in vitro, in a murine model, and in a cohort of 10 patients with CML,33 but has not been confirmed in subsequent retrospective and prospective studies.34–36
To date, there have been no reports of cure with imatinib, although patients may remain in remission for several months after interruption of therapy.37 Although the optimal frequency of hematologic and molecular monitoring once a stable dose of imatinib has been reached is unknown, it seems reasonable to perform a complete blood count every 3 months and molecular testing every 6 months in the absence of clinical signs suggestive of relapse. Screening for occult end organ involvement and/or evidence of drug toxicity, including clinical examination, routine chemistries, echocardiography, and pulmonary function testing, should also be performed every 6 months.
A single case of primary resistance to imatinib has been reported38 and secondary resistance appears to be rare, with only 7 cases reported in the literature to date.13,16,39–43 Most cases have been due to the appearance of a T674I mutation that is homologous to the T315I mutation that confers resistance to imatinib in CML, and recent data suggest that the low incidence of resistance may be due to a limited repertoire of possible mutations affecting the PDGFRA kinase domain.44 Second-line tyrosine kinase inhibitors, including nilotinib, sorafenib, and dasatinib, have in vitro activity against F/P, and nilotinib has been used successfully in one patient with imatinib resistant disease in the absence of a definable point mutation.43 Unfortunately, treatment of patients with imatinib resistance due to the T674I mutation has been ineffective to date, despite the availability of agents, such as sorafenib, that are effective against this mutation in vitro. In one case, outgrowth of a new pan-resistant clone after sorafenib treatment was the cause of the treatment failure.40 Nonmyeloablative allogeneic transplantation has been used successfully in HES45 and remains an option for the treatment of aggressive disease unresponsive to tyrosine kinase inhibitors.
CEL-NOS
CEL-NOS is the name given to unexplained eosinophilia > 1.5 × 109/L in the presence of > 2% peripheral blood or > 5% BM blasts, clonal eosinophils, or abnormal cytogenetics. To make a diagnosis of CEL-NOS, other myeloid neoplasms, clonal or phenotypically aberrant T cells, BCR/ABL, and abnormalities in PDGFRA, PDGFRB, and FGFR1 must be excluded. A variety of chromosomal abnormalities have been described in CEL-NOS, the most common of which is trisomy 8.46 Eosinophil clonality has also been demonstrated in the absence of cytogenetic abnormalities by HUMARA analysis in one female patient presenting with HES.47 Cases of CEL-NOS are exceedingly rare, but tend to be aggressive and unresponsive to therapy. IFN-α has been used successfully in a small number of cases, both alone48,49 and in combination with other agents.50 A short trial of imatinib 400 mg/d is also reasonable given the favorable side-effect profile, although the few cases of CEL-NOS treated with imatinib reported in the literature have been unresponsive. Finally, BM transplantation remains an option for CEL-NOS and should be considered early given the poor prognosis despite chemotherapy in most cases.
SM with eosinophilia
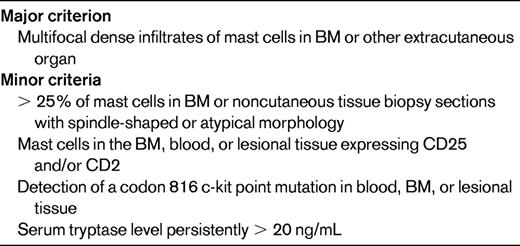
SM is a rare disorder characterized by an increased number of mast cells in the BM and/or other extracutaneous organs. The current WHO definition of SM requires the presence of 1 major + 1 minor criterion or 3 minor criteria (Table 3) and separates SM into disease variants based on the mast cell burden, involvement of non-mast cell lineages, and disease aggressiveness.3,51 The activating KIT mutation D816V is present in BM cells of 70%-93% of patients with SM,51,52 but is rarely detectable in the peripheral blood except in cases of mast cell leukemia. Other KIT mutations have been reported in SM, but are uncommon, representing < 3% of detectable abnormalities.51,52 Although SM with associated clonal hematological non-MC-lineage disease is a well-recognized SM variant, D816V KIT and F/P have not been detected in the same person to date.
Clinical and laboratory features
SM can occur at any age and is equally frequent in men and women. The clinical presentation is very variable, ranging from isolated BM involvement with minimal symptoms to rapidly fatal mast cell leukemia. Signs and symptoms are related to mast cell infiltration of tissues and mediator release, and classically include urticaria pigmentosa, flushing, abdominal pain, diarrhea, hepatosplenomegaly, lymphadenopathy, and bone pain. Constitutional symptoms, including fatigue, weight loss, fever, and night sweats are common, and anaphylaxis was seen in > 50% of patients in some series.53
Serum tryptase levels are elevated in nearly all patients with SM and are correlated with total mast cell burden.54 Additional laboratory abnormalities include anemia, thrombocytopenia, hypoalbuminemia, and elevated transaminases.55 BM examination typically shows a hypercellular BM with focal, dense, paratrabecular aggregates of atypical spindle-shaped mast cells and increased numbers of eosinophils and lymphocytes.56 Osteolytic or osteosclerotic changes in the bone trabeculae may accompany mast cell infiltration, and myelofibrosis is common, especially in advanced disease. Expression of CD25 and/or CD2 on CD117 (KIT)-positive mast cells can be demonstrated by immunohistochemistry or flow cytometry.51
Peripheral blood eosinophilia > 1.5 × 109/L is found in ∼ 15% of patients with F/P-negative SM29,55 and up to 50% of patients with D816V KIT-positive mast cell leukemia.52 This is likely due to the presence of the mutation in myeloid precursors. In fact, several studies have demonstrated decreased survival in patients with D816V KIT-positive SM and concomitant eosinophilia,57 perhaps due to clonal involvement at an earlier stage of myelopoiesis. Interestingly, however, the D816V KIT mutation was identified in both eosinophils and CD34+ hematopoietic stem cells in 30% of patients with mutation-positive SM, regardless of whether peripheral eosinophilia was present.52
Diagnosis
Diagnosis of SM requires careful morphologic and immunohistochemical analysis of the BM by an experienced pathologist. In addition, the presence of D816V KIT should be assessed in BM by RT-PCR with restriction fragment length polymorphism, peptide nucleic acid-mediated PCR, or allele-specific PCR.51 In the absence of a detectable D816V mutation and aggressive disease, additional analyses for mutations in KIT or other abnormalities should be performed, because the results may have therapeutic implications. As previously discussed, patients with peripheral eosinophilia > 1.5 × 109/L and BM mastocytosis present a unique diagnostic challenge due to overlapping clinical manifestations and BM findings in D816V KIT-positive SM and PDGFRA-associated myeloid neoplasms.52
Treatment
The treatment of patients with SM can be frustrating, because there is no curative therapy and no single agent has demonstrated success in a majority of patients. Treatment of indolent or smoldering SM is generally directed at reducing clinical symptoms. For more aggressive disease, several agents have been tried, with varying results. In a recent study examining the efficacy of chemotherapy in 108 F/P-negative patients with SM,58 response rates ranged from < 20% for imatinib and hydroxyurea to 53% and 55% for IFN-α and 2-chlorodeoxyadenosine, respectively. Complete responses were seen in only 2 patients, one who received IFN-α and one who was treated with 2-chlorodeoxyadenosine. The lack of response to imatinib is consistent with in vitro data demonstrating resistance of the D816V KIT mutation to imatinib therapy.59 It should be noted, however, that rare cases of SM with atypical KIT mutations have been shown to respond to imatinib.60,61 Dasatinib showed slightly better efficacy than imatinib in one trial.62 Newer agents, including PKC412, AMN107, and 17-AAG, have in vitro activity, but data on clinical efficacy are lacking. BM transplantation has been attempted in patients with advanced disease refractory to therapy, with discouraging results.63
Conclusions
Molecular diagnostics and targeted therapeutics are beginning to provide the necessary tools with which to reliably distinguish between the different myeloproliferative disorders associated with eosinophilia, including PDGFRA-associated myeloid neoplasms, CEL-NOS, and SM with peripheral eosinophilia. This has complicated the interpretation of results from prior clinical studies in which patients with disorders of differing etiologies were grouped together for analysis. As additional molecular markers are discovered and new subgroups of myeloproliferative HES are delineated, existing descriptions of clinical manifestations, prognosis, and response to treatment will need to be modified.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests. Off-label drug use: prednisone, hydroxyurea, IFN, mepolizumab, reslizumab, dasatinib, nilotinib, and sorafenib were all used off-label for the treatment of myeloproliferative HES.
Correspondence
Amy D. Klion, Laboratory of Parasitic Diseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Building 50/Room 6351, 50 South Dr, Bethesda, MD 20892; Phone: (301) 435-8903; Fax: (301) 451-2029; e-mail: aklion@niaid.nih.gov.