Abstract
A plethora of genetic abnormalities has been described in B-cell lymphomas, some of which arise when physiologic mechanisms involved in the generation of immunologic diversity go awry. Several different lymphoma types, such as follicular lymphoma (FL), mantle cell lymphoma (MCL), and Burkitt lymphoma (BL), are associated with hallmark translocations that occur as a consequence of these errors (t(14;18)(q32;q21), t(11;14)(q13;q32), and t(8;14)(q24;q32), respectively); however, none of these associations is absolute and none is completely diagnostically specific or sensitive. The advantages and limitations of a variety of different testing strategies in the 2 most common lymphomas, FL and diffuse large B-cell lymphoma (DLBCL), are reviewed herein, including an evaluation of the role of PCR-based approaches, FISH, and more nascent genomic technologies. The use of immunophenotypic strategies that may potentially provide, albeit imperfectly, more user-friendly surrogates for underlying genetic aberrations and cell-of-origin designations derived from gene-expression profiling analyses are also discussed. Finally, a newly designated category of lymphoma with features intermediate between DLBCL and BL is appraised, highlighting the central role of genetic analysis in this diagnostic gray zone.
Introduction
The contemporary classification of hematologic neoplasms relies upon the integration of clinical and pathologic features, with the latter encompassing morphology/histology, immunophenotyping, and genetic testing. Each of these 3 pathologic parameters has approximately equivalent relevance in current practice, but may assume more variable degrees of influence in different contexts. Genetic analysis includes several now standard technologies such as conventional karyotypic analysis, FISH, and PCR; however, such tools are neither always universally applied nor necessary. The availability of an ever-expanding menu of newer genetic technologies, including gene-expression profiling (GEP), array comparative genomic hybridization, methylation profiling, miRNA profiling, and deep sequencing, has led to an exponential growth in the discovery of the genetic underpinnings of a spectrum of leukemias and lymphomas. Attempts have been made to integrate many of these exciting newer findings into standard practice, but this has only met with modest success, largely because what applies in the research realm is not always easily translatable into robust, validated, and reproducible tests in day-to-day clinical diagnostics.1,2
For an ever-expanding number of neoplasms, genetic testing is mandatory, because some are defined and actually classified on the basis of the genetic aberration. In other neoplasms, genetic testing is not necessary for diagnosis but provides key prognostic information (eg, in chronic lymphocytic leukemia and plasma cell myeloma). Sometimes, such testing does not (yet) have an essential role. Amongst the numerous types of B-cell lymphomas, there are a few that are associated with characteristic hallmark translocations. These include Burkitt lymphoma (BL) and t(8;14)(q24;q32), or variants thereof, in which MYC is overexpressed; follicular lymphoma (FL) and t(14;18)(q32;q21), resulting in BCL2 overexpression; and mantle cell lymphoma (MCL) and t(11;14)(q13;q32), causing excessive expression of CCND1. All 3 of these translocations are a consequence of errors in the normally physiologic DNA breakage and rejoining that occurs in the IGH@ locus, either at the time of V(D)J recombination in the bone marrow and mediated by RAG1/RAG2 (recombinase-activating genes 1 and 2) in FL and MCL, or somatic hypermutation or class-switch recombination mediated by activation-induced cytosine deaminase in BL.3 However, unlike several myeloid neoplasms (typified by acute myeloid leukemias with recurrent genetic abnormalities and chronic myelogenous leukemia) and some B-cell lymphoblastic leukemias, in which translocations define specific entities, none of these 3 translocations is definitional; that is, none is either completely diagnostically specific or sensitive for the lymphoma with which it is linked. Therefore, for each lymphoma type, there are variable proportions of well-recognized cases that lack these hallmark translocations and yet they display numerous other defining and characteristic histologic and immunophenotypic features, allowing for their specific diagnosis. For example, it is possible to diagnose FL without a t(14;18) translocation, MCL without t(11;14), and even BL without t(8;14) or a variant translocation, albeit each with diminishing respective frequency.4 Up to 15% of FLs lack a t(14;18) translocation, as many as 10% of MCLs are negative for t(11;14), and the t(8;14) is absent in approximately 5% of BLs. Accordingly, the absence of these hallmark translocations does not, in isolation, exclude these diagnoses. As a corollary, not all t(14;18)-bearing lymphomas are FLs, because up to 25% of de novo diffuse large B-cell lymphomas (DLBCLs), excluding transformed FLs, harbor this translocation. Similarly, the t(11;14) translocation can occur in neoplasms that are not MCL, because it is present in a subset of plasma cell myelomas (which ought not to be confused with MCL) and t(8;14), classically linked with BL, is seen in some DLBCLs. Approximately 10% of de novo DLBCLs harbor MYC translocations. Because DLBCL is ∼ 15-20 times more common than BL, a MYC-positive lymphoma is actually more likely to be a DLBCL than a BL. However, it is probable that the same translocation has somewhat different biologic (and thus pathologic and clinical) connotations in different settings. Therefore, a MYC translocation in BL is considered to be a primary “driver” event, whereas in DLBCL, it is more typically associated with transformation/disease progression and thus is likely to be a secondary event.
With that backdrop in mind, the purpose of this short review is to highlight the role of a variety of “old” and “new” genetic approaches in the current diagnosis of 3 selected subtypes of B-cell lymphomas, to provide a rational framework for the choice of specific genetic tests in contemporary clinical practice, and to consider how nascent discoveries, or surrogates thereof, might (or might not) be applied in this setting.
FL
FL is a neoplasm of germinal center B cells that accounts for approximately 1/4 of all lymphomas diagnosed in North America and Europe (it is less common in Asia), making it the most common “indolent” lymphoma in these geographic locales. It is not the only B-cell lymphoma of germinal center origin. BL, approximately one-half of DLBCL cases, and probably Hodgkin lymphoma (in particular nodular lymphocyte–predominant Hodgkin lymphoma) also arise from this B-cell compartment. This frequent derivation from the germinal center is not altogether surprising, because it is here that B cells are subjected to physiologic DNA damage at 2 stages, as alluded to above (somatic hypermutation and class-switch recombination), which places them at risk for illegitimate and tumor-inducing mutations and translocations. However, the seminal translocation associated with FL, t(14;18), almost certainly occurs before the transformed cell(s) arrive in the germinal center, specifically in the bone marrow when VDJ recombination occurs. A large proportion of normal individuals harbor low numbers of circulating t(14;18)-positive cells, (apparently) without an increased risk for developing FL.5 Therefore, as is the situation in many other hematologic malignancies, the translocation appears to be necessary, but insufficient on its own, to induce neoplasia. It is only once these cells settle in the germinal center microenvironment that they are able to develop an overtly neoplastic phenotype, likely due to the permissive milieu and acquisition of additional genetic abnormalities. In fact, FL at presentation typically harbors not only the t(14;18) translocation, but also an average of 6 additional karyotypically detectable abnormalities, including 1p−, 6q−, +7, +12, and +X.6
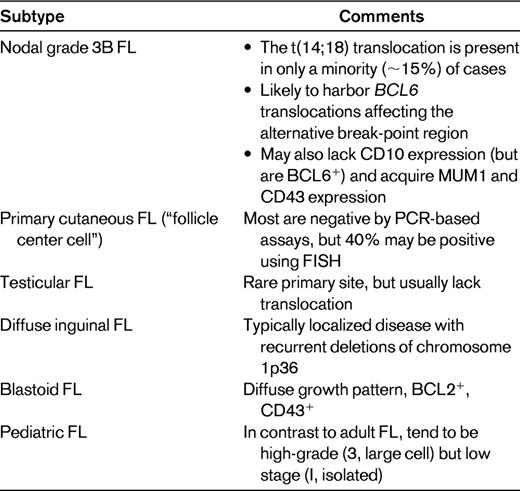
FL recapitulates much of the immunophenotype of normal germinal center B cells, namely the coexpression of CD10 and BCL6; however, unlike physiologic germinal center B cells, which are BCL2-negative, FL cells are characteristically BCL2-positive. This is a consequence of the t(14;18) translocation, which results in the dysregulated overexpression of the BCL2 gene, with the BCL2 protein allowing cells that are actually preprogrammed to die (from apoptosis) to survive. However, not all FLs harbor this translocation: ∼ 5%-15% of cases lack it. Nevertheless, if the classical histologic, morphologic, and immunophenotypic features are present, it is still possible to diagnose FL, noting that some (but not all) of these cases will not be BCL2-positive by immunohistochemistry. BCL2 may still be aberrantly overexpressed in some of these cases that lack the translocation, as a consequence of amplification of the BCL2 gene.7 Alternative oncogenic events in cases that lack BCL2 overexpression include translocations of BCL6, sometimes involving an alternative break-point region around the gene.8 Other frequent genetic lesions in FL are mutations of EZH2 (7%)9 and CREBBP (32%),10 although it is not clear if they are enriched in BCL2-negative cases. It is also possible, in some cases, to appear to lack detectable BCL2 protein expression despite the presence of an underlying t(14;18) translocation; this may occur when a point mutation in the BCL2 gene alters an epitope that would normally be recognized by the anti-BCL2 antibody used for immunohistochemistry.11 In this situation, the BCL2 protein is indeed overexpressed, but is not “seen” by the monoclonal antibody. There are variety of scenarios in which FLs, lacking t(14;18) and/or BCL2 protein expression (noting that these typically, but not invariably, accompany one another), are likely to be encountered. These include those occurring in extranodal sites (skin12 and testicles13 ), in specific age groups (pediatric14 ), and associated with morphologic variants (grade 3B, blastoid,15 and diffuse inguinal16 ; Table 1).
The “gold standard” for the detection of the t(14;18) translocation is classical karyotypic cytogenetic studies. However, fresh tissue is infrequently submitted for such analysis in most institutions. Alternative approaches to detect the translocation include PCR and FISH. Most standard PCR assays will, however, miss up to 25% of cytogenetically detectable translocations. This is because only 2 upstream BCL2 primers are typically used, one in the major break-point region and the other in the minor cluster region, which account for ∼ 60% and 15% of translocations, respectively. The addition of other primers, in particular those for the intermediate cluster region, will decrease the false negativity rate of PCR.17 However, FISH is even superior to this, because the probes used for this analysis are obviously able to span larger regions of DNA compared with short PCR primers.18 The immunoglobulin genes also seem to be a reasonable target for documenting clonality in this B-cell lymphoma. Standard IGH@ PCR has typically been problematic in FL, especially when using FR3 primers only, but this is now hopefully an infrequent occurrence currently, because of a high degree of false negativity as a consequence of ongoing somatic hypermutation. The use of IGK@ PCR usually overcomes this problem.19
In summary, it is possible to render a definitive diagnosis of FL in the absence of genetic testing, as long as the appropriate histologic and immunophenotypic features are present. However, in some scenarios, perhaps when there is limited tissue (as in needle core biopsies) and/or when the differential diagnosis might include a nodal marginal zone lymphoma in which there is follicular colonization, genetic testing may be valuable. It is here, and also when classical histologic and immunophenotypic findings are lacking, that the finding of a t(14;18) translocation can provide supportive evidence for a diagnosis of FL. In general, FISH rather than PCR is the preferred diagnostic modality absent the availability of fresh tissue for conventional chromosomal analysis. Finally, not all FLs are t(14;18)-positive (and one should be aware of the specific situations in which these cases are enriched) and not all t(14;18)-positive lymphomas are FLs.
DLBCL
DLBCL is the most common lymphoma in Western countries, accounting for approximately one-third of all adult lymphomas. Although historically lumped into one category both pathologically and clinically, it has become increasingly clear that DLBCL represents a heterogeneous admixture of quite different entities. This variability has been evident for some time at the clinical level, based upon different outcomes. Initial attempts to classify DLBCLs pathologically included a determination of whether the dominant cell type was centroblastic or immunoblastic. Although fairly strict microscopic criteria exist to distinguish the 2, this is not always feasible or reproducible in daily clinical practice; nevertheless, these 2 major morphologic subtypes remain inculcated in the contemporary (2008) World Health Organization (WHO) classification. Furthermore, as addressed below, there are recent data to suggest that the “low-tech” tool of microscopy may indeed still be of value. In addition to morphologic dissection of DLBCL, it has also become clear that the site of anatomic presentation is a key discriminator. Therefore, among the ∼ 40% of cases that present extranodally, it is necessary to separate out distinct subtypes such as those that arise in the CNS, mediastinum, skin, effusions, and intravascularly. Notwithstanding the importance of these histologic and clinical features that are germane to the contemporary classification of DLBCL, it has emerged that genetic tools appear particularly well suited to segregating different subtypes.
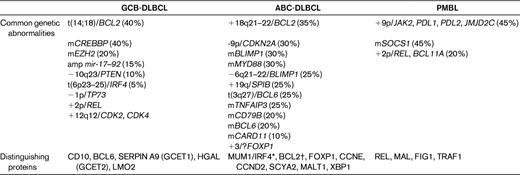
Single-gene approaches were initially applied to the categorization of DLBCL, with common targets for translocation and sometimes mutation being BCL6, BCL2, and MYC, in ∼ 40%, 25%, and 10% of cases, respectively.1 However, whereas certainly informative, they have been expanded upon via a variety of more global strategies, in particular GEP and array comparative genomic hybridization. Depending on the analytic approach (supervised vs unsupervised), a host of different types of DLBCL has been dissected using GEP. One classification that seems to have been adopted most frequently is that which distinguishes DLBCL into those that are of germinal center B-cell (GCB) or activated B-cell (ABC) origin20 ; a third form that separates out from this bimodal categorization is primary mediastinal DLBCL. GCB DLBCLs have consistently been shown to have a better prognosis than those derived from ABCs, and primary mediastinal DLBCLs usually have a slightly better outcome than GCB DLBCLs. An analysis of representative cell lines has shown that the expression profile of PMBL has more in common with classical Hodgkin lymphoma than with DLBCL, which is not altogether surprising given some of the shared clinical and pathologic features.20 These 3 major types, which are important to distinguish based upon their distinct prognoses and clinical features, have been investigated at a multitude of levels, and a plethora of intriguing genetic abnormalities has been identified (Table 2); however, only a few are tested for in contemporary practice.
GEP has been available for more than a decade now, and despite the promise that this technique might by now be evaluable in some user-friendly format that could be applied clinically, this still remains elusive. Related to this, there have been numerous attempts to develop surrogates that might convert the complexity of these approaches into more simple diagnostic strategies, which could be applied using existing robust technology. Several IHC algorithms have been proposed, with variable degrees of success in recapitulating the plethora of GEP data into a handful of antibody studies; these include those that variably test for the expression of CD10, BCL6, MUM1, GCET1, FOXP1, BCL2, and LMO2.21–23 With each algorithmic iteration, it appeared that there was increasing correlation with GEP, from ∼ 80% to ∼ 93%. However, it has also become clear that even though the technology of IHC is much more mature than that of GEP, it is not without standardization and reproducibility issues, suggesting that even such apparently simplified approaches have limitations and are not always reliable.24 Furthermore, whereas the initial IHC versus GEP studies alluded to above had a misclassification rate of only ∼ 7%-20%, more recent comparative analyses25 revealed that misclassification is even higher, especially for GCB subtypes, ranging from 30%-60%. Further, this study25 showed that whereas GEP continues to predict progression-free survival and overall survival based upon GCB versus ABC subtypes, none of the 5 IHC algorithms was able to do so. Therefore, stratification using IHC as a surrogate for GEP must, for now, be treated with reservation. At the same time, GEP or a simplified clinically adaptable incantation thereof, remains elusive in the contemporary evaluation of DLBCL in particular and lymphomas in general.
Alternative strategies that might be more adaptable to use in the clinical laboratory, and potentially applicable to formalin-fixed paraffin-embedded tissue, are quantitative RT-PCR26 and a quantitative ribonuclease protection assay.27 Only 6 genes (LMO2, BCL6, FN1, CCND2, SCYA3, and BCL2) were evaluated in the RT-PCR study: the first 3 are associated with a good prognosis and the last 3 with an adverse prognosis. The first 2 are GCB-associated genes and the last 3 ABC-associated genes. FN1, which encodes fibronectin, had been identified in other GEP studies as a gene associated with a favorable “lymph node” signature. The quantitative nuclease protection assay precludes the need for RNA isolation, reverse transcription, and amplification, and yet still enables measurement of gene expression. Whereas all of these attempts to develop tools that may provide substitutes for GEP are noteworthy, it is somewhat humbling that good old-fashioned microscopy, specifically in distinguishing immunoblastic morphology in DLBCL, currently retains robust, reproducible, and significant prognostic power that is in fact superior to the use of IHC markers.28
The contemporary relevance of the role of GEP and/or the use of IHC surrogates in the rituximab era is somewhat controversial. It has been proposed that BCL2-positive DLBCLs benefit from the addition of rituximab to CHOP chemotherapy, whereas BCL2-negative cases do not. Other studies suggest that this addition of rituximab to CHOP benefits BCL6-negative DLBCL, but not BCL6-positive cases. It therefore appears that ABC, but not GCB, DLBCLs benefit from the addition of rituximab. Some reports suggest that the use of rituximab seems to eliminate the prognostic differences between these 2 groups. However, other studies do not support this notion.
In summary, it is not necessary to perform any kind of genetic testing to render a diagnosis of DLBCL. However, it is clear that genetic factors and/or cell-of-origin data (GCB vs ABC) are key determinants of prognosis as well as of specific subtypes. Nevertheless, despite a decade of insightful investigation, none of the fascinating data emanating from GEP and other technologies have translated into routine clinical practice. Attempts to adopt IHC surrogates for GEP findings have met with only modest and controversial success. One genetic event in DLBCL, alluded to in the introduction and discussed in more detail below, namely MYC translocations, might be considered to be one of the more useful tests in DLBCL; however, it too does not (yet) play a role in diagnosis, but rather appears to have legitimate prognostic import.
B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and BL
The category of B-cell lymphoma, unclassifiable (referred to here as BCLU) was introduced by the WHO in 20084 to capture cases that have overlapping features of both DLBCL and BL, but fail to meet more rigorous criteria for either of these 2 more specifically defined entities. This group of lymphomas is mostly diagnosed in adults, and behaves aggressively with a tendency to be widespread at diagnosis, often involving the marrow and sometimes the peripheral blood.29 This category includes cases that might previously have been unsatisfactorily labeled as “atypical BL,” “non-BL,” or “Burkitt-like lymphoma”; use of these terms is no longer considered acceptable. These lymphomas may also be referred to as intermediate or gray zone lymphomas. One major intention of creating this rather awkwardly termed category is an attempt to maintain the “purity” of DLBCL and BL, and thus give pathologists a bona fide diagnostic category in which to place such less-well-defined cases. It is understandable that hematologist/oncologists may be frustrated by such terminology, but the intent is pure, because it is clear that clinical trials need to be undertaken to determine what the most effective therapy is for these lymphomas, noting that many do not typically do well with either prototypic DLBCL therapy or BL therapy. At the same time, pathologists are encouraged not to abuse this category, especially now that some latitude is allowed in the diagnosis of BL.
Consistent with normal pathology practice, BLCU cases are initially considered morphologically: for example, a DLBCL in which the cells are perhaps smaller than those typically seen or a highly proliferative lymphoma with a starry-sky pattern, in which the cells are somewhat larger than those seen with BL (Table 3). Others might have quite characteristic morphologic features but display an atypical immunophenotype; for example, a BL with strong BCL2 expression. However, it is perhaps at the genetic level that such cases display more characteristic findings (Table 3). Therefore, although a subset (∼ 40%) have MYC translocations, which are prototypically associated with BL, there are several important differences. First, the karyotype, if available, is likely to be complex in that a greater number of additional genetic abnormalities is typically seen (∼ 9 vs ∼ 2 in BL).30,31 Second, BCLUs are more likely to harbor BCL2 (and/or BCL6) translocations in addition to MYC translocations. Third, MYC translocations in BCLU may involve a non-IG partner; this is in contrast to BL, in which MYC translocations almost invariable involve an IG gene. However, not all DLBCLs with MYC translocations automatically qualify as BCLUs. Whereas BCLUs typically have morphologic (and immunophenotypic) clues as to the diagnosis, this is not usually the case in “generic” MYC-positive DLBCLs, which may be morphologically indistinguishable from those that lack this translocation. Such cases often behave more aggressively than MYC-negative DLBCLs.32,33
Double-hit B-cell lymphomas (DHLs), a somewhat imprecise term, include a spectrum of different, and characteristically clinically aggressive, lymphomas that do poorly with conventional therapy, including the use of high-intensity regimens.34 In the context of DLBCLs, they typically harbor a MYC translocation, together with another, most often a BCL2, translocation. Some are even triple-hit lymphomas, with the additional presence of BCL6 translocations. Histologically, they may resemble BCLUs, but they need not display overtly aggressive histologic features. Further, they typically have an immunophenotype of germinal center–derived B cells (ie, they usually coexpress CD10 and BCL6), together with BCL2 positivity and high Ki67 expression. However, whereas there is certainly some overlap between BCLUs and DHLs, they are not equivalent and not all BCLUs are DHLs (in fact, as noted, MYC translocations are evident in only ∼ 40% of BCLUs, whereas they are almost definitional in DHLs) and not all DHLs are BCLUs. Further investigation is necessary to dissect these categories and define their differences or reconcile their similarities.
In summary, it is acknowledged that the rather broadly designated category of BCLU is likely to evolve as it is further dissected, and it should be viewed as a temporary (and heterogeneous) holding category until such time as it is further defined. Nevertheless, genetic testing plays a central role in its designation. For now, unless a patient is not a candidate for aggressive therapy, it might be appropriate to consider screening all DLBCLs for not only MYC translocations, but also BCL2 and BCL6 translocations.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Adam Bagg, Department of Pathology & Laboratory Medicine, 3400 Spruce St, Philadelphia, PA 19104; Phone: (215) 662-6550; Fax: (215) 662-7529; e-mail: adambagg@mail.med.upenn.edu.