Abstract
Inherited platelet disorders (IPDs) comprise a heterogenous group of diseases that include defects in platelet function and disordered megakaryopoiesis. Some IPDs overlap as both defects in function and thrombopoiesis, resulting in both altered aggregation and/or secretion and thrombocytopenia. This review examines the key features of the presentation of IPDs in children and adults and presents a diagnostic algorithm for the evaluation of these patients. In addition, recent advances in our understanding of the pathophysiology of platelet disorders are addressed, with attention given to some of the novel genetic associations. Finally, treatment options and future therapies are briefly discussed.
Introduction: presentation of IPDs
Many of the severe congenital thrombocytopenias or platelet dysfunction disorders manifest early in life with bleeding symptoms, including mucosal bleeding such as epistaxis and gum bleeding and increased bruising with or without petechiae. Patients with inherited platelet disorders (IPDs) may also have life-threatening hemorrhaging in the gastrointestinal or genitourinary tracts or even intracranial hemorrhaging. Bleeding questionnaires have been developed for pediatric populations and for taking issues of diagnosing mild bleeding disorders in relatively hemostatically unchallenged individuals into consideration.1 However, some cases of congenital thrombocytopenia may not be detected until a screening complete blood count is done in late childhood or even as an adult, particularly in patients with disorders for which platelet function is relatively normal. In these patients, it may be difficult to differentiate between mild chronic, acquired thrombocytopenia, also called mild immune thrombocytopenia (ITP), and congenital thrombocytopenia. If a family history of thrombocytopenia is present, a comprehensive workup is warranted to avoid costly and potentially inappropriate therapies and to ensure appropriate follow-up of clinical complications, such as a predisposition to oncologic disorders, increased risk of renal disease, or sensorineural hearing loss.
Similar situations may exist for patients with inherited, mild platelet function defects, which may not manifest until a surgical or traumatic stress taxes the hemostatic system and results in significant bleeding. Many women with these mild disorders will present with menorrhagia, which may not be detected until she sees a gynecologist. Bleeding manifestations resemble those of type 1 or type 2 VWD—and the prevalence of IPDs may be as high as that seen in VWD—so diagnostic evaluation must rule out both diagnoses.
Patients with thrombocytopenia and associated features suggestive of some of the rarer but well-characterized disorders may also have features suggestive of those diseases; these include: reduced or delayed pigmentation (Hermansky Pudlak syndrome); personal or family history of hearing loss, renal impairment, or cataracts (MYH9-related disorders); eczema, infection, and small platelets (Wiskott Aldrich syndrome [WAS] and X-linked thrombocytopenia); limb abnormalities (thrombocytopenia absent radii [TAR]); and familial predisposition to leukemia/lymphoma (RUNX1 mutations).
Advances in our understanding of genetics and pathophysiology
Whereas most IPDs are individually rare, being found in 1 in 104–6 persons, in aggregate they are likely to be more common than typically presumed; the true prevalence, however, is not known.2,3 In addition, IPDs have been important in providing tools to dissect the complex lives of platelets from megakaryopoiesis to platelet apoptosis. Through the characterization of IPDs, we have learned much about the role of platelet surface proteins such as glycoprotein IIb (GPIIb)/IIIa (associated with Glanzmann thrombasthenia [GT]),4 GPIb/IX/V complex (associated with Bernard Soulier Syndrome [BSS]),5 GPVI,6 GPIa-IIa,7 P2Y12 receptor,8 and the thromboxane receptor.9–11 Studies of families with platelet function defects have led to the definition of signaling pathway defects, including mutations of cyclooxygenase-112 or thromboxane A synthetase.13 These studies have also led to the discovery of disorders of platelet granule formation, such as grey platelet syndrome (GPS), alpha granule deficiency, delta granule deficiency, or of content, such as Quebec platelet syndrome and Montreal platelet syndrome. Studies of MYH9-related disorders led to a further understanding of the importance of cytoskeletal proteins in thrombopoiesis. Finally, disordered megakaryopoiesis, as seen in familial platelet disorder with predisposition to acute myeloid leukemia, congenital amegakaryocytic thrombocytopenia (CAMT), X-linked thrombocytopenia/thalassemia, and amegakaryocytic thrombocytopenia with radio-ulnar synostosis (ATRUS), have led to our increased understanding of the role of thrombopoietin (TPO) and key transcription factors in the maintenance of platelet count and BM integrity.
Many of the mild platelet disorders show an autosomal-dominant inheritance pattern, so family history is key in making these diagnoses. More significant thrombocytopenia and/or platelet dysfunction can be seen in disorders with autosomal-recessive or X-linked inheritance patterns, which may be more common among populations in which consanguinity is common or in association with some ethnicities with a high prevalence of a particular IPD. In addition, timing of onset of bleeding symptoms can help to differentiate between acquired and inherited disorders, because most patients with IPDs will begin to manifest bleeding in childhood.
Diagnostic algorithm
Initial evaluation must include careful history with specific attention to ethnicity and consanguinity, bleeding history with and without trauma or surgery, and associated medical complications consistent with some of the more common IPDs. An evaluation of the family history is especially important for children because they may have had few hemostatic challenges. Particular attention in this population should be paid to umbilical stump bleeding, bleeding after circumcision, cephalohematoma, recurrent petechiae and excessive purpura for developmental age, conjunctival bleeding, and macroscopic hematuria. Several bleeding scores have been validated, including one for VWD in pediatrics that has been applied to bleeding in platelet disorders in general.1,14 This is particularly germane to IPDs as well because the bleeding manifestations can be quite similar.
The first and most common laboratory tests performed to assess platelet function are the platelet count and peripheral blood smear. These can guide further evaluations and are the first step in a diagnostic algorithm (Figure 1). Careful evaluation of platelet count and morphology on the peripheral smear is important because many cell counters will underestimate the platelet count in the setting of macrothrombocytopenia or microthrombocytopenia, and on an automated cell counter the mean platelet volume may be inaccurate in these settings.15 Additional cell morphology that may point to a diagnosis on the peripheral smear includes: pale, large platelets in GPS; Dohle-like inclusions in the WBC in MYH9-related disorders; and abnormal RBC morphology suggesting GATA-1 mutations.

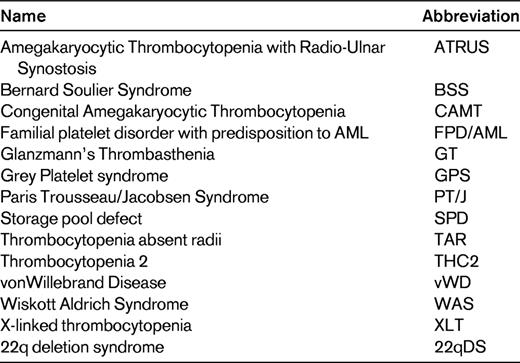
Diagnostic algorithm for IPDs. See Table 1 for abbreviations. (Adapted with permission from Israels et al.15 )
Normal platelet counts
If the platelet count is normal and a bleeding disorder is suspected, evaluation of platelet function is indicated. Because manifestations of mild platelet function defects can be similar to those of VWD, evaluation for VWD needs to occur either before or concurrently with evaluation of platelet function.16 Despite efforts to develop smaller-volume or less-time-intensive screening assays for platelet disorders, light transmission aggregometry remains the single most common and effective method of assessing platelet function. Recently published guidelines should help to standardize this testing.17 Unfortunately, full platelet aggregation panels require relatively large volumes of blood, making them difficult to perform in smaller pediatric patients. In addition, nonspecific abnormal platelet aggregometry should be repeated on fresh blood samples in an experienced laboratory to document persistent abnormalities and to minimize the chances of assigning a diagnosis of bleeding disorder due to testing difficulties. These factors combined make it necessary for smaller children to return several times to complete the diagnostic workup. Minor aggregation abnormalities are frequently seen in the general population, especially weak responses to epinephrine or ADP, and may represent variant physiology rather than disease.18,19
Specific platelet aggregation defects can establish a diagnosis: absent response to all agonists except ristocetin should prompt confirmatory flow cytometric testing for reduced/absent αΙΙb/β3 expression and mutational analysis to identify the genetic defects of GT. A markedly decreased or absent response to ADP indicates a P2Y12 receptor defect, and mutational analysis or flow cytometry can confirm this after a careful medication history to exclude drug-induced platelet aggregation defect. Decreased secondary aggregation to ADP and epinephrine and decreased aggregation in response to collagen suggests a platelet storage defect, which can be confirmed in most patients by an ATP- or serotonin-release assay. Electron microscopy in an experienced laboratory can help to distinguish whether the defect is due to the absence of granules or to the ineffective release of granule contents. Increased agglutination with low concentrations of ristocetin suggests type 2B or platelet-type VWD (confirmed by VWD testing and mixing studies and mutation analysis), whereas absent response to ristocetin suggests BSS, which can be confirmed by flow cytometry and mutation analysis.
The least progress has been made in the subgroup of patients without these specific platelet aggregation defects, but with abnormal platelet aggregometry. After careful review of the history to rule out drug effects and assessment for renal and liver disease, mild abnormalities may indicate an IPD. Several family members with the same aggregation defect might help to establish the heritability of the disorder. Careful examination of the blood smear for morphologic changes suggestive of myelodysplastic syndrome should be performed and, if indicated, a BM examination should be arranged. Genome-wide and whole-exon sequencing studies are likely to help in the future by allowing for the identification of genes important in the regulation of platelet function in humans.
Finally, in patients with normal aggregometry but mucocutaneous bleeding, consideration should be given to an evaluation for Scott syndrome, which is caused by defective scramblase activity and results in the inability to expose phosphatidylserine on the surface of platelets (or apoptotic cells).20 This disorder can by diagnosed by the lack of Annexin V binding to activated platelets. Recently, mutations in associated proteins, including transmembrane protein TMEM16F, have also been shown to result in deficient scramblase activity.21,22
Thrombocytopenia
If the patient has thrombocytopenia, the diagnostic evaluation should be guided by platelet size as follows.
Small platelets.
Small platelet size in a male patient should prompt genetic evaluation for mutations in the X-linked WAS protein and investigation for associated cellular and humoral immunodeficiency, eczema, and recurrent infections. Megakaryocytes on BM studies are typically normal. Both WAS and X-linked thrombocytopenia were recently reviewed by Thrasher.23
Normal platelets.
Normal platelet size may indicate a defect in megakaryopoiesis. Investigation in those cases in which ITP is not the leading diagnosis should include a BM examination (eg, congenital onset, poor/absent response to ITP therapy, suspected chronic lymphocytic leukemia, or systemic symptoms/signs of disease).24 Whereas thrombocytopenia in neonates is fairly common, rare inherited conditions such as CAMT, ATRUS, thrombocytopenia absent radius (TAR), and Paris Trousseau/Jacobsen syndrome present at birth with severe thrombocytopenia. Plasma TPO levels may help in differentiating these disorders from acquired thrombocytopenia due to perinatal or prenatal factors,25 particularly with regard to CAMT, in which the plasma TPO levels can be more than 10-fold higher than normal. ATRUS and TAR both present with characteristic bony abnormalities, including radioulnar synostosis and/or absent radii, so that upper extremity X-ray examination may be helpful in more subtle bony abnormalities. CAMT can be confirmed with analysis of the TPO-receptor gene, c-Mpl.26,27 All of these patients will all have severe thrombocytopenia persisting beyond the neonatal period, but patients with TAR classically show partial platelet count recovery over the first few years of life.26 In contrast, CAMT does not show improved platelet counts over time and many patients will progress to BM failure.25
An inherited, autosomal-dominant thrombocytopenia with normal platelet size called thrombocytopenia 2 has been mapped to chromosome 10p11.1-p12.28 Noris et. al recently described mutations in ANKRD26 (within the region of linkage mapping), a gene of unknown function. It is known that ANKRD26 is expressed in megakaryocytes and is overexpressed in some tumors, resulting in thrombocytopenia in 12 additional families. This describes more fully the thrombocytopenia 2 phenotype and suggests that there may be an association with acute leukemia in affected pedigrees.29,30
Finally, familial platelet disorder with predisposition to acute myeloid leukemia is an autosomal-dominant inherited disorder with mild to moderate thrombocytopenia, mild to moderate platelet function abnormalities, and a 35% risk of leukemia caused by haploinsufficiency of RUNX1.31
Large platelets.
Large platelets are classically seen in several inherited disorders of platelets. However, the most common cause of large platelets, particularly in the pediatric population, is acquired immune thrombocytopenia (ITP). Most of these patients will present with a history of acute onset of significant mucocutaneous bleeding that is a distinct change from their baseline. However, when children present with “ITP” that is refractory to all treatments and/or there is a family history of thrombocytopenia, an inherited, congenital thrombocytopenia must be considered.
Platelet-type or type-2B VWD must be considered in patients with chronic thrombocytopenia and, often, large platelets.32–34 Testing for VWF antigen and activity levels and low-dose ristocetin-induced platelet aggregation can establish this diagnosis. In addition, mutation analysis of VWF and GPIbα can identify activating mutations responsible for this disorder.35,36
Familial studies have identified mutations in MYH9, which codes for the heavy chain of nonmuscle myosin IIA (NMMHC-IIA) and is known to be responsible for the May-Hegglin anomaly and Fechtner, Sebastian, and Epstein syndromes.37 The presence of Dohle-like bodies in leukocytes on the peripheral smear may be subtle, but the likelihood of detection can be enhanced by immunofluorescence studies for aggregates of NMMHC-IIA and mutation analysis is clinically available to detect changes in MYH9. Family history of associated nephritis, hearing loss, or cataracts may also be suggestive, although children often do not manifest these findings and hearing loss can be quite subtle, requiring formal audiometry for early detection (reviewed in Althaus and Greinacher38 ).
GPS often presents with large, pale platelets on the peripheral smear. Further, aggregometry may show decreased platelet aggregation with collagen and/or thrombin and electron microscopy should show the absence of mature alpha granules. A recent study used single-nucleotide polymorphism array analysis to narrow the previously reported 9-mb region on chromosome 3p21 to a 1.7-mb region on the same chromosome.39,40
Large, granular platelets in a patient with moderate to severe bleeding and thrombocytopenia suggest BSS, which can be diagnosed by flow cytometric analysis of the surface expression of the GPIb/IX/V complex in most patients. The majority of patients have autosomal-recessive inheritance patterns and the heterozygous individuals are unaffected.3 However, autosomal-dominant mutations resulting in a mild phenotype have also been reported and may be the most common cause of mild macrothrombocytopenia.41–43 Mutation analysis of the GPIBα, GPIBβ, and GPIX genes is also clinically available and can aid in the genetic counseling of affected families and in establishing the diagnosis in patients with a present but dysfunctional receptor complex.3
Paris Trousseau/Jacobsen syndrome patients have deletion of Fli1, a key transcription factor important in megakaryopoiesis on chromosome 11q23. These neonates have associated cardiac and other defects and developmental delays.44,45 Characteristic platelet abnormalities include giant platelets with large alpha granules visible on electron microscopy.46 These patients also have qualitative and quantitative platelet defects and may improve with age.
Finally, in those patients without significant bleeding and mild macrothrombocytopenia with decreased expression of the GPIb/IX/V complex, benign Mediterranean macrothrombocytopenia has been reported to be due to heterozygosity of mutations in the complex.47 In addition, thought should be given to whether investigation for 22q deletion syndrome (22qDS) is clinically indicated. 22qDS is relatively common, affecting at least 1 in 4000 live births.48 Associated symptoms include cardiac anomalies, hypocalcemia, and immune defects, but these may be subtle and many adult carriers of the deletion are now being detected because of mutation analysis of children and parents.48 Patients with the classic deletion not only have deletion of the TBX1 gene thought to be primarily responsible for the cardiac and velofacial anomalies associated with the syndrome, but also have deletion of GPIBB. Patients with 22qDS have decreased platelet counts (compared with children without 22qDS), increased mean platelet volume, and decreased expression of the GPIb/IX/V complex on the surface of platelets.49,50
Treatment
Patients with IPDs may have significant bleeding. The most severe disorders (GT, BSS, CAMT, and WAS) may be associated with life-threatening hemorrhaging,2,3,23,25 and platelet transfusion may be required to stop significant bleeding. Because of the risk of alloimmunization, it may be important to perform HLA typing in patients with these severe disorders and transfuse them with HLA-matched platelets if available. Leukocyte-depleted transfusions should always be used; however, platelet transfusion should never be withheld in these patients in the setting of significant, potentially life-threatening bleeding just because of fear of alloimmunization. General physicians understand the use of platelet transfusions in the face of significant thrombocytopenia, but may require guidance in its use in the face of a normal platelet count.
Local measures
Other measures are important in the management of bleeding in IPD patients so as to avoid platelet transfusions. All patients should all be counseled to avoid antiplatelet medications. Hormonal contraception may decrease menorrhagia in women with platelet disorders.51 Proper dental hygiene from infancy is important to preempt invasive dental procedures later in life. Education of the risks of bleeding and its impact on elective surgeries such as wisdom teeth extraction is often needed for parents and medical caregivers.
Proper compression of the nose/nasal bridge during epistaxis (and venipuncture sites) is the frontline in the management of these most frequent complications, especially in pediatric patients. Topical sealants can also be used to manage gum bleeding and epistaxis.52 Conservative management with topical sealants and/or pressure is often sufficient. Cauterization may be effective in short-term management, but may result in more frequent and recalcitrant nosebleeds in the long term and should be avoided if possible. Proper oral hygiene is also important to prevent gum disease, which will increase the risk of mouth bleeding.
Antifibrinolytic therapy
Use of aminocaproic acid or tranexamic acid in the setting of mucocutaneous bleeding, menorrhagia, or gastrointestinal bleeding has been reported in many patients with both inherited platelet dysfunction and thrombocytopenias.53 These medications may decrease the need for routine platelet transfusions and should be considered early in bleeding episodes.
Desmopressin
Desmopressin (1-desamino-8-D-arginine vasopressin) can shorten the bleeding time in patients with platelet function defects,54 likely due to the stimulation of the release of endothelial cell VWF, which allows enhanced platelet adhesion to vessel walls. However, it is not as effective in patients with dense granule deficiency. Therefore, in more severe platelet disorders such as BSS/GT, desmopressin is unlikely to be effective, but in some platelet disorders (particularly with normal dense granules) it may be transfusion sparing.55
rFVIIa
Recombinant activated factor VII (rFVIIa) is often used in patients with bleeding refractory to conventional therapy. However, almost all of the data to support the use of rFVIIa in platelet disorders are anecdotal or based on small case series. Poon et al used rFVIIa as prophylaxis in patients with GT undergoing surgery and showed that it was effective in 29 of 31 patients at a dose of 80-120 μg/kg every 1.5-3 hours for 3 doses or until hemostasis was achieved.56 This regimen has also been used in patients with other platelet disorders.57,58 However, in established bleeding, rFVIIa has shown less efficacy, particularly in GT, for which only 64% of patients achieved hemostasis with rFVIIa only in the registry report by Poon.59 Another study showed that the use of rFVIIa did not significantly reduce the need for platelet transfusions.58 Given the proposed mechanism of action of rFVIIa, it is more likely to be effective when given with platelet transfusion.
TPO receptor agonists
Several reports have been published examining the utility of TPO-receptor agonists in the management of patients with inherited thrombocytopenias.60,61 These have shown some efficacy in increasing the platelet count in some disorders, including MYH9-related diseases and CAMT, but have had variable success in CAMT, as would be expected given its pathophysiology.25
Future therapies
Several groups of investigators have been working toward generating novel therapies for patients with IPDs. Wilcox et al have been investigating the potential for gene therapy of specific defects, and have shown correction of GT in both murine and canine models.62 BM transplantation has been performed in a patients with WAS and GT, resulting in normalization of platelet function and megakaryopoiesis.63,64 There have been similar efforts in GT, but it most such patients have a near-normal lifestyle and the cost-benefit of such therapy needs better evaluation. Use of autologous platelet-rich clots has also been reported to be effective in skin biopsy and dental extraction in patients with type 2 VWD, platelet-type VWD, and GT.65 Perhaps most interesting are recent efforts to generate megakaryocytes and/or platelets ex vivo from induced pluripotent stem (iPS) cells.66 These studies offer the possibility of reprogramming one's own hematopoietic cells or fibroblasts to generate iPS cells that can then be corrected for the platelet defect, allowing for autotransfusion of HLA-identical, genetically corrected platelets. Alternatively, banks of iPS cells generating ex vivo platelets may someday allow for the transfusion of HLA-matched or even one's own platelets to a degree not feasible due to the current reliance on platelet donations.
Summary
In summary, whereas they are individually rare, in aggregate, IPDs are common causes of mucocutaneous bleeding. The descriptions of underlying defects are slowly being defined and therapeutic intervention is also improving. In patients with family history suggesting a heritable condition, or in persons with significant, lifelong bleeding histories, comprehensive evaluation for the presence of IPDs is important and significantly affects prognosis and treatment.
Disclosures
Conflict-of-interest disclosure: The author has received honoraria from Cangene. Off-label drug use: None disclosed.
Correspondence
Michele P. Lambert, MD, The Children's Hospital of Philadelphia, 3615 Civic Center Blvd, ARC, Rm 316G, Philadelphia, PA 19104; Phone: (215) 590-4667; Fax: (267) 426-5476; e-mail: lambertm@email.chop.edu.