Abstract
Myelodysplastic syndromes (MDS) are a heterogenous group of hematologic malignancies characterized by clonal expansion of BM myeloid cells with impaired differentiation. The identification of recurrent mutations in MDS samples has led to new insights into the pathophysiology of these disorders. Of particular interest is the recent recognition that genes involved in the regulation of histone function (EZH2, ASXL1, and UTX) and DNA methylation (DNMT3A, IDH1/IDH2, and TET2) are recurrently mutated in MDS, providing an important link between genetic and epigenetic alterations in this disease. The mechanism by which these mutated genes contribute to disease pathogenesis is an active area of research, with a current focus on which downstream target genes may be affected. Recent advances from sequencing studies suggest that multiple mutations are required for MDS initiation and progression to acute myeloid leukemia (AML). The past several years have yielded many new insights, but the complete genetic landscape of MDS is not yet known. Moreover, few (if any) of the findings are sufficiently robust to be incorporated into routine clinical practice at this time. Additional studies will be required to understand the prognostic implications of these mutations for treatment response, progression to AML, and survival.
Introduction
Ten years ago, the genetics of myelodysplastic syndromes (MDS) was largely defined by cytogenetic abnormalities. Although our understanding of MDS still lags behind other myeloid malignancies such as chronic myeloid leukemia, acute myeloid leukemia (AML), and myeloproliferative neoplasm (MPN), much has been learned in recent years and the pace of discovery is accelerating. A detailed understanding of MDS genetics is essential, because it provides insight into MDS biology, will help to refine prognostic models, and can guide treatment decisions, including therapies targeted to specific genetic lesions. Technology development has been a major driver of recent discoveries in MDS genetics as the field has moved from “low-resolution genome scans” using conventional cytogenetics and candidate gene resequencing to array-based karyotyping, genome-wide mRNA/miRNA expression profiling, and now genome-wide sequence analysis. Because of space constraints, only recent advances in MDS genetics will be cited here (including seminal and comprehensive studies from the past 5 years). Readers are encouraged to consult several recent reviews for further information.1,2
Karyotype abnormalities in MDS and gene haploinsufficiency
The karyotype of BM cells remains one of the most important prognostic markers for MDS and is a key component in both the International Prognostic Scoring System and the World Health Organization Classification-Based Scoring System. Copy number alterations (deletions and amplifications) of all or part of chromosomes 5 (36% of patients), 7 (21% of patients), 8 (16% of patients), and 20 (7% of patients) are among the most common cytogenetic abnormalities observed in MDS, and altered regions are thought to harbor important genes that contribute to MDS pathogenesis.3 Single nucleotide polymorphism (SNP) arrays have allowed investigators to fine-map copy number alteration break points, identify microdeletions below the resolution of cytogenetics, and discover regions of copy number–neutral loss of heterozygosity (uniparental disomies or UPDs). Detection of subcytogenetic copy number alterations using SNP array complements standard metaphase cytogenetics and provides independent prognostic value for MDS patients.4 Detection of UPD in MDS BM cells has been a valuable marker for identifying segments that harbor biallelic mutations in genes, including EZH2, CBL, TET2, and TP53.5–8
An interstitial deletion of one copy of chromosome 5q (either alone or in the setting of complex cytogenetics) is the most common cytogenetic change in de novo MDS.3 Several studies have mapped the commonly deleted regions (CDRs) on 5q, and this resulted in the identification of a proximal region on 5q31.2 that is associated with a high risk for transformation to AML and a distal region on 5q33.1 that is associated with the 5q− syndrome and a lower risk of AML transformation (reviewed in Bejar et al1 ). Candidate gene-resequencing studies have not identified a biallelic gene mutation on either interval, suggesting that gene haploinsufficiency is the genetic mechanism contributing to MDS initiation and progression.9
Using an shRNA screen in primary human CD34+ cells, haploinsufficiency of RPS14 on the distal 5q33 region was found to contribute to the abnormal erythroid differentiation and apoptosis that is commonly observed in patients with the 5q− syndrome.10 A mouse model that is haploinsufficient for Rps14 and 7 other genes is characterized by macrocytic anemia, further supporting the hypothesis that RPS14 haploinsufficiency is an important MDS-initiating event.11 It was then shown that up-regulation of TP53 is essential for the RPS14 haploinsufficiency phenotype.11,12 However, haploinsufficiency of RPS14 does not recapitulate all of the features of the 5q− syndrome, including thrombocytosis, suggesting that haploinsufficiency of additional gene(s) on 5q33 may cooperate with RPS14 loss. Indeed, haploinsufficiency of miR-145 and miR-146, both located on the 5q33 CDR, leads to thrombocytosis in vivo.13
Additional features of MDS may be explained by haploinsufficiency of genes on the 5q31 interval (EGR1, CTNNA1, and HSPA9) and 5q genes located outside of both CDRs (NPM1 and APC). Haploinsufficiency of Egr1 enhances stem-cell self-renewal and increases the risk of N-ethyl-N-nitrosourea–induced leukemia in mice.14,15 Hypermethylation of the residual nondeleted CTNNA1 allele is associated with del(5q)–transformed AML in humans.16 Reduced expression of HSPA9 in primary human CD34+ cells results in delayed erythroid maturation and increased apoptosis in vitro and decreased hematopoietic progenitors in mice.17 Npm+/− mice develop erythroid macrocytosis, megakaryocytic dysplasia, and are predisposed to myeloid malignancies (AML and MPNs) at 10-24 months of age.18 Finally, Apc+/− mice have alterations in their stem cell compartment with concomitant changes in cell cycle and apoptosis which may be relevant for MDS.19,20 Collectively, these results implicate multigene haploinsufficiency as the dominant genetic mechanism caused by chromosome 5 deletions and suggest that gene haploinsufficiency may be the mechanism contributing to disease pathogenesis for other common deletions in MDS.
Several CDRs have been mapped for chromosome 7q deletions (7q21, 7q22, 7q32-33, and 7q35-36) (reviewed in Bejar et al1 ). Identification of the critical genes in these intervals has been lacking until recently with the identification of mutations in EZH2 located on 7q36 in patients with MDS.6,7 No definitive causal gene has been identified for chromosome 20q deletions (although ASXL1 is located on 20q) or trisomy 8.
Although the mechanism underlying the generation of interstitial deletions is not known, the recent description of chromosomal shattering and rejoining of segments has been observed in many types of cancers, including leukemia.21 This process, called chromothripsis, provides an explanation for complex deletions observed in MDS samples (Figure 1), and may be another mechanism creating genetic diversity during MDS initiation and progression to secondary AML (sAML). Whole-genome sequencing will be a valuable tool to better define these complex genetic alterations in MDS genomes.

Chromothripsis of chromosome 5 in MDS. Multiple regions of DNA copy number (CN) loss are detectable by SNP arrays comparing a tumor sample (MDS-derived sAML) with the patient's normal DNA (obtained from a skin biopsy) (top panel). Whole-genome sequence data of the tumor/normal pair demonstrates loss of heterozygosity (LOH) at the regions of copy number loss (middle panel). Multiple inversions (INV) and intrachromosomal translocations (ITX) involving retained segments flanking the deletions (DEL) are computationally predicted to be rearranged from the whole-genome sequence data (bottom panel; T.G. and M.J.W., unpublished data).
Chromothripsis of chromosome 5 in MDS. Multiple regions of DNA copy number (CN) loss are detectable by SNP arrays comparing a tumor sample (MDS-derived sAML) with the patient's normal DNA (obtained from a skin biopsy) (top panel). Whole-genome sequence data of the tumor/normal pair demonstrates loss of heterozygosity (LOH) at the regions of copy number loss (middle panel). Multiple inversions (INV) and intrachromosomal translocations (ITX) involving retained segments flanking the deletions (DEL) are computationally predicted to be rearranged from the whole-genome sequence data (bottom panel; T.G. and M.J.W., unpublished data).
Mutations in regulators of DNA methylation
Alterations in DNA methylation have been implicated in the pathogenesis of MDS.22 This is supported by observations that the global methylation pattern of cytosine residues in CpG dinucleotide sequences is different between normal and MDS BM cells22 and that there is clinical benefit to patients with MDS when treated with cytosine analog drugs that interfere with methylation (5-azacytidine and 5-aza-2′-deoxycytidine). Cytosine methylation status influences gene transcription, which may contribute to the altered growth and differentiation of MDS cells. There is now genetic evidence that DNA methylation is important in MDS, because several genes that regulate cytosine methylation are somatically mutated in MDS genomes (DNMT3A, TET2, and IDH1/IDH2).
Methylation at the 5′ position of cytosine in CpG dinucleotides is mediated by DNA methyltransferases (DNMTs; Figure 2). DNMT3A and DNMT3B are the dominant DNMTs involved in de novo methylation and DNMT1 maintains hemimethylated DNA during replication. 5′-methylcytosine (5mC) can be further modified by a group of 3 paralogous Ten Eleven Translocation (TET) proteins (TET1, TET2, and TET3), that are alpha-ketoglutarate (αKG) and Fe(II)-dependent oxygenases that catalyze the conversion of 5mC to 5′-hydroxymethylcytosine (5hmC).23 5hmC appears to be a short-lived intermediary that may lead to demethylation of cytosine, although its precise fate remains unknown. The main source of αKG comes from the conversion of isocitrate to αKG by isocitrate dehydrogenase 1 (IDH1) in the cytosol and IDH2 in the mitochondria and TET function is reduced when αKG levels are limiting in a cell.23 Based on their known functions, it is predicted that DNMT3A, TET2, or IDH1/IDH2 gene mutations alter cytosine methylation, and ultimately gene transcription, by affecting either 5mC or 5hmC levels.

Genetic alterations of epigenetic pathways in MDS. The normal function of selected factors important for histone modification and DNA methylation is depicted. Left panel: In most cases, the biological consequences of mutations in these pathways is not yet well-established. Trimethylation (me3) of lysine 27 (K27) on the carboxyterminal tail of histone H3, a nucleosome component, is associated with transcriptional repression. ASXL1 is a polycomb repressive complex protein that maintains the repressive state. Both ASXL1 and EZH2 (encoding an H3K27 methyltransferase) are frequently mutated in MDS. Rare mutations (deletions) have also been identified in MDS samples in H3K27 demethylase enzymes, including UTX and other JmjC domain–containing proteins. Right panel: Cytosine methylation in CpG islands is associated with transcriptional repression. DNMT3A is a de novo DNA methyltransferase that converts unmethylated cytosine to 5mC. 5mC is converted to 5hmC by the TET proteins in the presence of αKG generated by the IDH enzymes. DNMT3A, TET2, IDH1, and IDH2 are all recurrently mutated in MDS samples.
Genetic alterations of epigenetic pathways in MDS. The normal function of selected factors important for histone modification and DNA methylation is depicted. Left panel: In most cases, the biological consequences of mutations in these pathways is not yet well-established. Trimethylation (me3) of lysine 27 (K27) on the carboxyterminal tail of histone H3, a nucleosome component, is associated with transcriptional repression. ASXL1 is a polycomb repressive complex protein that maintains the repressive state. Both ASXL1 and EZH2 (encoding an H3K27 methyltransferase) are frequently mutated in MDS. Rare mutations (deletions) have also been identified in MDS samples in H3K27 demethylase enzymes, including UTX and other JmjC domain–containing proteins. Right panel: Cytosine methylation in CpG islands is associated with transcriptional repression. DNMT3A is a de novo DNA methyltransferase that converts unmethylated cytosine to 5mC. 5mC is converted to 5hmC by the TET proteins in the presence of αKG generated by the IDH enzymes. DNMT3A, TET2, IDH1, and IDH2 are all recurrently mutated in MDS samples.
Mutations in DNMT3A were first discovered in AML and were subsequently identified in up to 8% of de novo MDS samples.24–27 The most frequent mutation in AML and MDS is a heterozygous missense mutation that converts arginine to histidine at position 882 (R882H). This mutation reduces the methyltransferase activity of DNMT3A in vitro,24 but it has not been studied in combination with the wild-type allele, and it is unknown if the mutation results in a gain or loss of function. Compound heterozygous, frame shift, and nonsense mutations occur, suggesting that loss-of-function mutations do exist. Mutations were found in all subtypes of MDS and patients with mutations have a worse overall, event-free, and AML-free survival.26 Recent studies of murine Dnmt3a−/− hematopoietic stem cells suggest that loss of Dnmt3a leads to a competitive advantage over wild-type cells,28 providing evidence that these mutations may contribute to clonal dominance. Consistent with this finding, the allele frequency of DNMT3A mutations was ∼ 50% for both DNA and RNA isolated from unfractionated BM samples from MDS patients (suggesting that 100% of cells harbor a heterozygous mutation), even when the myeloblast count was normal.26
Deletions and UPDs spanning chromosome 4q24 were observed in patients with AML, MDS, and MPN.29,30 Sequencing of TET2 (located in a 4q24 microdeletion) identified missense, frame shift, and nonsense somatic mutations in myeloid cancers, including 11%-26% of patients with MDS, 37%-44% of patients with MDS/MPN, and 11%-24% of patients with sAML.29–34 Hemizygous and compound heterozygous mutations are common, and many mutations are predicted to produce loss of function. Indeed, several missense mutations are associated with impaired 5hmC production and reduced 5hmC levels in vitro and in vivo, respectively.35 Although it is predicted that genomes with TET2 mutations would be hypermethylated, both global hypermethylation36 and hypomethylation35 have been observed. These conflicting results may represent platform differences or they may indicate that the critical subset of genes/loci that are affected have not yet been identified. No consistent impact of TET2 mutations on survival has been observed in MDS.31,34
Mutations in IDH1 were first described in gliomas and were then independently identified in an AML genome using whole-genome sequencing.37 IDH1 and IDH2 mutations in AML are highly associated with intermediate-risk cytogenetics and a normal karyotype. Mutations predominantly involve codons R132 of IDH1 and R140 and R172 of IDH2. IDH1/IDH2 mutations are rare in MDS (4%-11%), but may be more common in sAML (8%-10%).38–40 Patients with IDH1 mutations have inferior overall survival compared with nonmutated patients.38 Heterozygous IDH1/IDH2 mutations result in the production of 2-hydroxyglutarate, which is dependent on the presence of the wild-type allele.41 This is consistent with the identification of only heterozygous mutations in patients. IDH1/IDH2 and TET2 mutations are mutually exclusive in AML and share a similar hypermethylation pattern in patient samples.36 This is likely due to the ability of mutant IDH1/IDH2 to inhibit 5hmC production by TET2.36 Additional enzymes, including certain histone demethylases, are also dependent on αKG for their catalytic activity and may be affected by 2-hydroxyglutarate production.36
Mutations affecting histone function
Histone proteins contribute to regulation of gene expression by dynamically organizing DNA into zones of active (“open”) and inactive (“closed”) chromatin. This process is regulated in part by a complex series of posttranslational modifications (including acetylation, methylation, and others) to histone tails (Figure 2). These modifications affect the recruitment of transcriptional regulators (eg, transcription factors, corepressors, and coactivators) and the histone-modifying enzymes themselves (eg, histone acetyltransferases, deacetylases, methyltransferases, and demethylases). Trimethylation of the lysine at position 27 in histone H3 (H3K27) typically results in reduced gene expression, suggesting that mutations that decrease H3K27 methylation activate transcription.
Recently, recurrent mutations in several genes encoding histone regulators have been identified in MDS patients. The first described involve EZH2 (enhancer of zeste homolog 2), a polycomb group protein that methylates histones H3 (at K27) and H1 (at K26), generally resulting in transcriptional repression. EZH2 point mutations (including missense, nonsense, frame shift, splice site, and deletions) were detected in 2%-6% of MDS patients.6,7,42 The mutations cluster in the c-terminal catalytic SET (suppressor of variegation, enhancer of zeste, trithorax) domain, but were also found in the C-rich and SANT (switch-defective protein 3, adaptor 2, nuclear receptor corepressor, transcription factor) domains. In roughly half of the cases, the mutations are hemizygous or biallelic (via microdeletions of 7q or UPD, respectively).6,7,42 The EZH2 Y641 codon that is frequency mutated in follicular and diffuse large B-cell lymphomas does not appear to be a common target of mutation in MDS. In vitro experiments suggest that the EZH2 mutations found in MDS patients are predominantly loss of function (resulting in reduced H3K27 trimethylation)6,42 and may be associated with relatively poor survival.42 The H3K27 trimethylation mark recruits DNMTs to sites of de novo methylation, providing a link between two repressive epigenetic pathways.
UTX, an X-linked polycomb gene containing a JmjC domain, encodes an H3K27 demethylase. Rare deletions of UTX32 and other JmjC family members—but so far no point mutations—have been reported in MDS. Finally, the ASXL1 (additional sex comb-like 1) gene encodes a member of the enhancer of trithorax and polycomb group of proteins that maintain transcriptional activation or repression in different cellular contexts. Mutations in ASXL1 have been found in 11%-15% of MDS patients.32,43 All of the described mutations are protein-truncating frame shifts predicted to delete the C-terminal PHD domain.
At this time, there are insufficient data to draw firm conclusions about the potential impact of these mutations on survival or evolution to sAML. Defining the biological consequences and the patterns of cooperating or mutually exclusive mutations in genes affecting histone regulation or DNA methylation in MDS is also very much a work in progress.
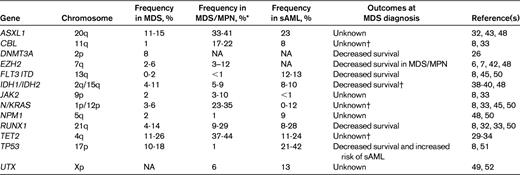
Secondary MDS and MDS/AML
The incidence of MDS is increased in individuals who are exposed to genotoxins (eg, ionizing radiation and alkylator chemotherapy) or who inherit predisposing genetic variants. The spectrum of acquired genetic lesions in de novo and secondary MDS is similar, although the mutational frequencies are different. For example, TP53 mutations are relatively infrequent in de novo MDS (Table 1), but are more common in therapy-related MDS (25%-30%).44 RUNX1 and N/KRAS mutations are also more common in therapy-related MDS (15%-30% and 10%-12%, respectively),44,45 compared with de novo MDS (Table 1). Balanced translocations (including rearrangements at the RUNX1 and MLL loci) are also infrequent in de novo MDS, but are more common in radiation-induced MDS.44 Unbalanced loss of material from chromosomes 5 and 7, a frequent event in de novo MDS (see above), is even more common in alkylator-associated MDS.44
Rare Mendelian disorders are associated with a strong predisposition to MDS. These include the FDP (familial disorder of platelets) syndrome (OMIM 601399), caused by inherited RUNX1 mutations and a recently described syndrome associated with GATA2 variants.46 MDS may also arise as a consequence of an inherited BM failure syndrome. The causal genes for many of these syndromes have been identified (eg, DKC1, TERC, and others for Dyskeratosis congenita; FANC genes for Fanconi anemia, ELANE and others for severe congenital neutropenia; and SBDS in Shwachman-Diamond syndrome).
Approximately 1/3 of MDS cases transform to sAML. New genetic lesions (molecular or cytogenetic) are detectable at the sAML stage in more than half of these cases.47 The genetic progression factors reported so far are not sAML specific (ie, they occur in other patients with MDS or AML), but there are differences in frequency. In general, the frequency of known single-gene mutations is lower in MDS-derived sAML compared with de novo AML, and is lower still in MDS at diagnosis. For example, FLT3-ITD mutations are uncommon in de novo MDS, but are enriched in sAML (Table 1), though not to the extent that they occur in de novo AML (∼ 20%).25 NPMc mutations are also detectable at a lower frequency in MDS and sAML (Table 1) compared with de novo AML (∼ 23%).25 Serial studies in individual patients have shown that FLT3-ITD, RUNX1, and NPMc mutations arise between MDS diagnosis and evolution to sAML in at least half of cases.47
MDS/MPN Overlap Syndromes
In the most recent World Health Organization system, chronic myelomonocytic leukemia (CMML) was reclassified as an MDS/MPN overlap syndrome, together with several other myeloid malignancies characterized by various degrees of dysplasia and hyperproliferation. Not surprisingly, mutations that tend to be overrepresented in these disorders (compared with de novo MDS) affect cell proliferation and survival pathways. For example, the CBL proteins are adapter molecules that modulate cytokine receptor signaling through the Ras pathway. CBL mutations are much more frequent (∼ 20%) in CMML compared with MDS (< 5%) and are mutually exclusive with N/KRAS, RUNX1, and JAK2 mutations.8,33 Most of the CBL mutations are missense and often (in > 80% of cases) become homozygous in CMML samples as a result of UPD11q.8
Mutations affecting the polycomb repressive complex are also common in MDS/MPN patients. EZH2 is mutated in 3%-12% of MDS/MPN cases.6,42,48 A preliminary report found missense mutations in UTX at a similar frequency (∼ 6%) in MDS/MPN,49 while ASXL1 mutations are more common (33%-41%) in CMML.32,43,48 EZH2 and ASXL1 mutations co-occur in CMML about as often as would be expected by chance.48
Conclusions
Thanks to the efforts of a growing international community of MDS researchers, there has been enormous growth in our understanding of MDS genetics. Most of the insights have come from careful analysis of paired tumor/normal samples from well-phenotyped patients. In several cases, the recognition of recurrent cytogenetic abnormalities (eg, deletions and UPDs) provided clues leading to the discovery of novel, recurrent single-gene mutations. Although molecular or cytogenetic abnormalities can now be detected in a majority of patients with MDS or MDS/MPN overlap syndromes, the full landscape of genetic abnormalities in these diseases is not yet known.
Recurring mutations in genes encoding regulators of the epigenome (Figure 2) represent a major new paradigm in MDS genetics that has emerged over the past 2 years. Important research priorities in the near term include defining the prevalence of these mutations in all subtypes of MDS and other myeloid malignancies, screening other genes in these pathways for mutations, and defining the biological consequences of these mutations. Much work needs to be done to determine what clinical impact these mutations may have on survival, AML progression, and response to therapy, particularly with drugs that target histone modifications and/or DNA methylation. Currently, it would be premature to advocate for mutational profiling of MDS samples in routine clinical practice. This recommendation must be revisited once new findings have been replicated and extended into large cohorts of MDS patients in which treatment history, comorbidity, and other covariates are annotated. The current phase of unbiased whole-exome or whole-genome sequencing is revealing abnormalities in new genes and pathways in these diseases. This information is likely to add clarity to diagnosis and classification, improve risk stratification, and help to guide treatment decisions for patients with MDS.
Note added in proof: While this chapter was in preparation, the following paper was published. Bejar R, Stevenson K, Abdel-Wahab O, et al. Clincial effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364(26):2496-2506.
Disclosures
Conflict-of-interest disclosures: The authors declare no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Timothy Graubert, Division of Oncology, Washington University School of Medicine, 660 South Euclid Ave, Campus Box 8007, St Louis, MO 63110; Phone: (314) 747-4437; Fax: (314) 362-9333; e-mail: graubert@medicine.wustl.edu.