Abstract
In 1882, Philippe Gaucher described a 32-year-old woman with massive splenomegaly and unusually large cells in the spleen, which he called a “primary epithelioma of the spleen.” The systemic nature and inheritance of the disease and its variants involving the viscera and CNS were described over the next century. The delineation of the causal enzymatic defects, genetics, molecular pathology, and genomics have provided pathogenic insights into the phenotypic spectrum and the bases for development of specific therapies for what is now known as Gaucher disease. As a prototype, the clinically and economically successful intracellular enzyme therapy provided the impetus for the expansion of similar research and therapeutic developments for other lysosomal storage diseases (LSDs) and orphan diseases, including Fabry, Pompe, and Niemann-Pick diseases, as well as several mucopolysaccharidoses. Continuing studies of such LSDs, which occur as a group in more than 7000 live births, have revealed the complex molecular interdigitation with the autophagy and apoptotic pathways and proteostasis and the impact of disruptions of the lysosomal/autophagy and proteostasis systems on more common diseases has been recognized. Examples include age-related neurodegenerative diseases (eg, Parkinson disease and Gaucher disease), idiopathic hypertrophic myocardiopathies, stroke and renal failure (eg, Fabry disease), and Nonalcoholic Fatty Liver Disease/Nonalcoholic SteatoHepatitis (NAFLD/NASH) and atherosclerosis (eg, lysosomal acid lipase deficiencies). Although perceived as rare, the availability of treatment and the impact of the LSDs on more common diseases require their integration into routine clinical practice.
Introduction
In 1882, Philippe Gaucher described a 32-year-old woman with massive splenomegaly and unusually large cells in the spleen, which he called a “primary epithelioma of the spleen.” The systemic nature of this disease, its inheritance, and variants that involve the viscera and CNS were described over the next century.1–3 The delineation of the causal enzymatic, genetic, and molecular pathology and genomics have provided pathologic insights into the phenotypic spectrum and bases for the development of specific therapies for what is now known as Gaucher disease. This disease was the first lysosomal storage disease (LSD) described and has become a prototype for the clinical description and phenotypic variability of more than 50 LSDs. These LSDs include several variants of Gaucher, Fabry, Pompe, and Niemann-Pick diseases, as well as several mucopolysaccharidoses and other disorders caused by defective function of the more than 300 lysosomal enzymes or lysosomal membrane proteins.4 These diseases manifest as a variety of visceral and CNS diseases. In aggregate, they occur with a frequency of 1 in 7000 to 1 in 3000 live births. Herein, the focus will be on Gaucher disease because of its principal involvement of hematopoietic-derived cellular systems and because it is a prototype for other similar diseases, including Niemann-Pick disease type A and B, the lysosomal acid lipase deficiency disorders, Wolman disease, and cholesteryl ester storage disease.5,6 The phenotypic variation and the importance of specific hematopoietic cell type involvement will be emphasized as the basis for the development specific enzyme therapy (ET) and other approaches to the treatment of these disorders. Finally, the impact on the disruptions that these diseases cause on the lysosomal/autophagy systems and proteostasis will be extrapolated to more common disorders and the role of these systems in neurodegenerative and hematologic malignancies.
Manifestations and clinical phenotypes
A guiding principle in the understanding of LSDs, and Gaucher disease in particular, has been the identification of specific primary cell types that are involved in the disease process. In the visceral variants of Gaucher disease, the major involved organs include the spleen, liver, and lungs and the cortical and bone marrow compartments.7 These organs are involved specifically because the principally affected cells are macrophage derived. The primary enzyme defect, acid β-glucosidase, leads to the accumulation of an excess indigestible substrate, glucosylceramide (Figure 1), in organs that contain significant numbers of macrophage-lineage cells. Recent studies have also shown the involvement of most myeloid- and lymphoid-derived cells and their elaboration of numerous cytokines and chemokines.8 The primary source for the accumulated glucosylceramide in visceral organs is the turnover of senescent blood-formed element membranes that contain glycosphingolipids, including leukocytes, erythrocytes, and platelets.9 With the development of hepatomegaly, splenomegaly, and BM expansion, the disease process becomes an accelerating vicious cycle of accumulating material that activates macrophages and leads to the production of additional macrophages and accumulating Gaucher cells in the various tissues. Although the primary storage material, glucosylceramide, leads to classical and alternatively activated macrophages, the exact mechanism by which this activation and the subsequent production of numerous cytokines and chemokines has not been defined clearly. Therefore, the tissues involved in the visceral Gaucher disease variant called type 1 or non-neuronopathic disease have accumulations of Gaucher cells and other activated APCs with inflammation, production of IFNs and IL-4 pathway–mediated cytokines and chemokines, the development of fibrotic changes, and changes in the space occupying the lesion caused by the progressive accumulated Gaucher cells. The pro- and anti-inflammatory effects lead to irreversible tissue damage and the variably progressive nature of the disease. Classically, Gaucher disease type 1 leads to hepatosplenomegaly, BM expansion, cortical bone mineral losses, and hypoproductive- and consumptive-based anemia and thrombocytopenia.7 Less commonly, additional organs, particularly the lungs, can be involved, but are usually not the major manifestations of the disease. When the lungs are involved by infiltrative disease, hypertension, or arteriovenous shunting, this is life-threatening.10 The variably progressive nature of the hepatosplenomegaly and the chronic bone disease can lead to severe disability in affected patients.

Glycosphingolipid (GSL) lysosomal degradative pathway. The sequential removal of glycosidic residues from the ceramide backbone of GSLs by specific lysosomal enzymes is shown. Glucosylceramide is the penultimate GSL in this pathway and the enzyme acid β-glucosidase has defective function in Gaucher disease. Each of the other enzyme deficiencies in this pathway are associated with specific LSDs (eg, α-galactosidase A in Fabry disease).
Glycosphingolipid (GSL) lysosomal degradative pathway. The sequential removal of glycosidic residues from the ceramide backbone of GSLs by specific lysosomal enzymes is shown. Glucosylceramide is the penultimate GSL in this pathway and the enzyme acid β-glucosidase has defective function in Gaucher disease. Each of the other enzyme deficiencies in this pathway are associated with specific LSDs (eg, α-galactosidase A in Fabry disease).
These primary and secondary events due to APC activation and enhanced inflammatory responses are the results of defective function of a single enzyme, acid β-glucosidase (EC3.2.1.45).11 This enzymatic deficiency is not absolute, in that every patient with Gaucher disease, irrespective of severity, has some amount of residual enzyme activity present in all nucleated cell types. Indeed, the absence of acid β-glucosidase in humans or mice is lethal in the preterm or neonatal periods.7
Molecular genetics and enzymology of Gaucher disease
The defects in acid β-glucosidase function result from more than 350 different mutations in the GBA1 gene, including point mutations, insertions/deletions, and rearrangements.7 The point mutant alleles lead to the production of acid β-glucosidases with functional, kinetic, trafficking, and/or stability defects and their resultant decrease in lysosomal function and increased accumulation glucosylceramide. A GBA1 pseudogene is highly homologous with GBA1 and is approximately 34 kb downstream from the functional gene.7 Recurrent gene rearrangements and/or gene conversions lead to pseudogene changes appearing in Gaucher disease–causative GBA1 alleles.
There are several common gene mutations associated with Gaucher disease in the Western world. A mutation termed N370S is very common and originates from the Ashkenazi Jewish population. Gaucher disease type 1 has a frequency of approximately 1 in 800 to 1 in 1000 persons in that population.7 The N370S allele has been distributed to other populations in Europe, North America, and Israel, where it is the most common Gaucher disease allele. In addition, this mutant allele confers, in the homoallelic or heteroallelic (ie, 2 different GBA1 mutant alleles) states, protection against the early-life development of primary (insert missing characters) disease. The most common mutant GBA1 allele worldwide is L444P, which results from recurrent gene-conversion events with the pseudogene. Homozygosity for L444P is found almost exclusively in patients with primary neuronopathic disease. In addition, these gene conversions may result in isolated L444P alleles or alleles that contain L444P and one to several pseudogene-derived additional mutations.7 These multiply substituted alleles are null and produce no functional acid β-glucosidase.
Unlike soluble lysosomal proteins, the membrane-associated acid β-glucosidase is not trafficked to the lysosome via the mannose 6-phosphate receptor (M6PR) system during intracellular synthesis.12 Instead, lysosomal integral membrane protein 2 (LIMP2) is the receptor for a peptide sequence on acid β-glucosidase for delivery to the lysosome. Once bound in the endoplasmic reticulum to LIMP2, acid β-glucosidase is transported through the Golgi apparatus to the trans-Golgi network, where it is dissociated from LIMP2 via a pH decrease and becomes membrane associated in the lysosome.13 Normally, acid β-glucosidase is not secreted, unlike many soluble lysosomal proteins.14 Most of the other lysosomal enzymes that are soluble are trafficked to the lysosome by the M6PR system. All lysosomal enzymes are glycoproteins with various oligosaccharide modifications as a result of remodeling with passage through the Golgi.15
In Gaucher disease, the acid β-glucosidase–defective functions and substrate excesses lead not only to disruptions in macrophage/APC function, but also to enhanced pro- and anti-inflammatory cytokine and chemokine production. The subsequent downstream molecular pathologic changes in tissues lead to fibrosis and other repair defects. This altered inflammatory environment is also seen in variants of Gaucher disease that involve the CNS. These variants, termed type 2 (acute neuronopathic) and type 3 (subacute neuronopathic), present early in life and have progressive neuronopathic involvement.7 Although these variants have been classified as distinct entities, they represent a continuum of neuronopathic involvement, with type 2 having rapidly progressing CNS deterioration and visceral disease and type 3 manifesting more highly variable neuronopathic and visceral progression.16 In contrast to the visceral disease in types 1, 2, and 3, the neuronopathic involvement in types 2 and 3 Gaucher disease results, not from phagocytosis of blood-formed elements, but from the inability to degrade and dispose of glucosylceramide and a derivative neurotoxin, glucosyl sphingosine, which are produced endogenously in CNS neuronal cells. The consequent neuronal dysfunction elicits apoptotic or other programmed cell death “eat me” signals for the activation of astroglial and microglial cells. Therefore, the CNS disease is a primary loss of neurons with subsequent neuroinflammatory effects, whereas the visceral disease is primarily macrophage/inflammatory cell initiated and mediated.17,18 Importantly for therapy, the blood-brain barrier hinders the entry of IV-administered enzymes into the CNS. In addition, unlike the diseases that result from lysosomal enzymes, which can be normally secreted and taken up by neighboring cells, Gaucher disease types 2 and 3 are not amenable to hematopoietic stem cell transplantation approaches that depend on metabolic cross-correction.19
Genotype/phenotype correlations
With the development of large LSD registries (> 6000 for Gaucher disease and > 4000 for Fabry disease), the relationships of genotypes and phenotypes for prognostic significance have evolved. The N370S allele confers protection against the early onset of primary (insert missing characters) involvement in Gaucher disease, whereas the L444P homozygosity highly predisposes to the development of early-life (insert missing characters) disease.7 Many other combinations of mutant alleles lead to substantially reduced enzymatic activity with in cells and variations in the phenotype. As might be expected, a lower amount of residual activity in specific cells is correlated with more rapidly progressing disease. A threshold will eventually be crossed and early-life (insert missing characters) disease will manifest. Therefore, the genotypes of patients with Gaucher disease can provide guidance as to the nature of disease progression, the expected result of therapy (see “Therapies”), and the prognostic significance for families as a basis for genetic counseling and family planning. This prognostic significance should be placed against the background of the occasional, significant sibling variation in the manifestations of Gaucher disease. Therefore, absolute prognosis cannot be given, but, among siblings, the general trend is toward similarity of involvement (with some exceptions). Details of the genotype/phenotype correlations, the pathologic involvement, and other aspects of the enzymology and molecular pathology of LSDs are available in comprehensive reviews.7,20
Therapies
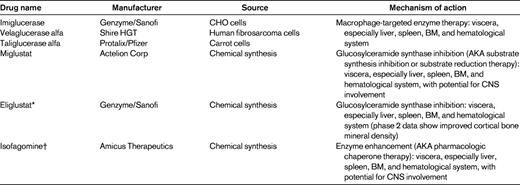
Gaucher disease is the prototype for ET for LSDs and several other disorders. The initial studies conducted at the National Institutes of Health by Dr Roscoe Brady's group indicated that specific modifications of the oligosaccharide chains of the purified natural enzyme would be required for targeting to specific tissues and for therapeutic effect21 (Table 1). These studies demonstrated the essential need for specific cellular targeting and dosing to provide significant therapeutic effects.22 As indicated in the previous section, for newly synthesized acid β-glucosidase inside the cell, LIMP2 is the receptor for lysosomal targeting; this is not true for delivery of enzyme from outside of the cell to the lysosome. Because of the presence on macrophages of a specific mannose receptor,23 first natural placental and then recombinant acid β-glucosidases were modified to expose terminal mannosyl residues of the oligosaccharide chains. This led to the preferential uptake of IV-administered acid β-glucosidase into the affected macrophages and the degradation of the stored glucosylceramide. The concept of specific tissue targeting has been applied to ET for the other LSDs and indicates the need for such preferential targeting, because misdirection of the enzyme to alternative tissues can lead to a blunting or absence of the therapeutic response.24
Using the macrophage mannose receptor as a “Trojan horse” for the delivery of the enzyme to the lysosomes, 3 ETs have been developed for Gaucher disease. Two of these, imiglucerase (Genzyme) and velaglucerase alfa (Shire HGT) are currently approved worldwide for the treatment of significantly involved patients with Gaucher disease type 1. These are produced, respectively, in Chinese hamster ovary cells or a human fibrosarcoma cell line. Another product, taliglucerase alfa (Protalix/Pfizer), was approved for the treatment of adults (> 18 years of age) by the United States Food and Drug Administration (FDA) on May 3, 2012. This enzyme is produced in transgenic carrot cells.25 Each of these enzymes has different oligosaccharide modifications that may affect their interactions, uptake, and delivery to different cell types. This has been studied thoroughly with imiglucerase and velaglucerase alfa in mouse models of Gaucher disease, and the essential equivalency of these 2 enzymes has been demonstrated.26 Clinical trials in which equal doses of either enzyme are used have also shown high clinical similarity. Based on limited data, taliglucerase appears to have some similarity to the other 2 enzymes. Therefore, ET by the IV administration of recombinant-produced enzymes that are macrophage-preferentially targeted has become the standard of care for Gaucher disease type 1. The results demonstrate a significant dose-response effect that can be used to guide appropriate dosing in various types of patients for reducing major manifestations of the disease. The liver, spleen, and hematologic abnormalities return to normalcy in 1-4 years, whereas the bone may take many years to have significant responses regarding cortical bone density. IV-administered ET has similar effectiveness for the visceral manifestations of Gaucher disease types 2 and 3, but does not have effects on the neuronopathic involvement. Overall, the results of ET have been rather remarkable and, in many cases, lifesaving.
Substrate synthesis inhibition therapy (AKA substrate reduction therapy) is an alternative approach first proposed by Norman Radin.27 This approach decreases the synthesis of glucosylceramide or other offending storage materials by inhibiting the appropriate synthetic enzyme (eg, glucosylceramide synthase), with a resultant decreased production of this offending lipid and the ability of the residual enzyme activity to reestablish a new steady state. This approach was first tried with deoxynojirimycin derivatives and, in particular, the N-butyl derivative that led to FDA and European Medicines Evaluation Agency (EMA) approval of miglustat (ie, Zavesca and Actelion).28 Clinical trials demonstrated effects on the visceral organs in Gaucher disease type 1 with some diminution in hepatosplenomegaly and hematologic findings. However, there were substantial adverse events related to the use of miglustat, in particular, significant diarrhea. Although diet modification has had some effect on this side effect, the acceptance for this drug in the United States has been low. In addition, there are other more controversial potential adverse events for miglustat, including tremor and paresthesias.29 Newer agents have been developed based on the original work of Radin. For example, eliglustat (Genzyme) is now being tested. Instead of being similar to the glucose moiety of glucosylceramide, as are the deoxynojirimycins, eliglustat has structural properties similar to the ceramide moiety. Phase 2 trials demonstrated a high efficacy in patients who are naive to any therapy and, significantly, showed reversal of hepatosplenomegaly and improvement in anemia and thrombocytopenia. Unexpectedly, substantial and relatively prompt increases were observed in bone mineral density in several patients.30 Phase 3 trials are under way and data analyses should begin in 2013. The obvious advantage of these 2 agents is their oral availability and their potential to cross the brain-blood barrier and have CNS effects. Miglustat does cross the blood-brain barrier and has some effects in animal models on CNS glycosphingolipids. Eliglustat is a p-glycoprotein substrate that is pumped out of the CNS and never reaches therapeutic levels. Based on the structure of eliglustat, newer compounds are under investigation that may have enhanced CNS effects.
A third approach to therapy for Gaucher disease was termed pharmacologic chaperone therapy, but the term enzyme enhancement therapy (EET) is preferable because it more accurately reflects the mechanistic bases. This approach is based on the counterintuitive notion that highly potent and specific competitive inhibitors would bind to mutant enzymes in the endoplasmic reticulum or other parts of the protein synthesis system and reconform or conform enzymes to a more active state by improving either their stability or their fundamental kinetic properties or both.31 The need would be to increase the residual mutant enzyme level above the threshold to provide therapeutic effects. In tissue culture and some animal model systems, positive effects have been obtained on enzymatic function.32 The clinical trials with isofagomine for Gaucher disease type 1 was stopped because there were no general clinical effects. An advantage of this approach is that the oral agents that can penetrate the blood-brain barrier and have activity in the CNS. There are continuing trials with EET for Fabry and Pompe diseases.
The therapeutic approach that is just on the horizon is the use of induced pluripotent stem cell–derived stem cell transplantation. For Gaucher disease and other lysosomal disorders, wild-type donor BM transplantation has been used because monocytes from the peripheral blood can migrate across the blood-brain barrier and become CNS microglial cells that could affect metabolic cross-correction. For Gaucher disease, bone marrow or stem cell transplantation has not been effective for the CNS disease because of the lack of secretable enzyme. Induced pluripotent stem cells are an attractive alternative for generating either hematologic progenitor cells or neural progenitor cells for direct cellular and/or enhanced gene therapy.
Several of the other LSDs for which ET is used generally do not have primary hematologic involvement, include Fabry disease (agalsidase alfa and beta), mucopolysaccharidosis types I (laronidase) and II (idursulfase), and Pompe disease (glycogenosis type II and alglucosidase alfa). Several of the approaches described herein have been and are available for the treatment of these diseases; in particular ET. ET for Niemann-Pick B disease (a non-neuronopathic acid sphingomyelinase deficiency) is currently in clinical trials. In addition, clinical trials are ongoing for EET in Fabry disease and Pompe disease. The results of these trials should become available in the near future and indicate their potential for clinical impact. Similarly, the successes in ET for all of these diseases has stimulated the development of newer approaches for ET for more directed or enhanced ligand binding to target tissues in the various diseases (eg, Pompe disease and directed muscle-preferential uptake).
Finally, the involvement of the lysosomal/autophagy system in the development of chronic diseases of adulthood, particularly of the neurodegenerative diseases, has come to the fore recently. The pivotal studies by Nichols et al33 and Sidransky et al34 demonstrated that Parkinson disease patients are carriers of GBA1 mutations (heterozygotes). Indeed, GBA1 mutations are the leading modifier of Parkinson disease manifestations, and are correlated with earlier onset and potentially more rapidly progressive neurodegenerative disease than occurs in those patients without such mutations. The molecular bases are being investigated, but there is a clear relationship between the accumulation of α-synuclein and the diminished activity of acid β-glucosidase in specific parts of the CNS that lead to the manifestations of Parkinson disease.35 This finding has led to speculation that other lysosomal enzymes in which there is partial disruption or abnormalities of the lysosomal/autophagy system may be equally important for other neurodegenerative diseases such as Alzheimer disease. The future evaluation of potential therapies for LSDs may be extended as interventions to ameliorate other such disorders.
Disclosures
Conflict-of-interest disclosure: The author has received research funding from Genzyme/Sanofi and Shire HGT, has consulted for Genzyme/Sanofi, and has received honoraria from Genzyme/Sanofi and Shire HGT. Off-label drug use: None disclosed.
Correspondence
Gregory A. Grabowski, MD, A. Graeme Mitchell Chair in Human Genetics, Professor, and Director, Division of Human Genetics, Cincinnati Children's Hospital Medical Center, 3333 Burnet Ave, MLC 4006, Cincinnati, OH 45229-3039; Phone: 513-636-7290; Fax: 513-636-2261; e-mail: greg.grabowski@cchmc.org.