Abstract
Atypical hemolytic uremic syndrome (aHUS) is a rare syndrome of hemolysis, thrombocytopenia, and renal insufficiency. Genetic mutations in the alternate pathway of complement are well recognized as the cause in more than 60% of patients affected by this thrombotic microangiopathy. The identification of aHUS as a disease of the alternate pathway of complement enables directed therapeutic intervention both in the acute and chronic setting and may include one or all of the following: plasma therapy, complement blockade, and liver transplantation. Because aHUS shares many of the presenting characteristics of the other thrombotic microangiopathies, and confirmatory genetic results are not available at the time of presentation, the diagnosis relies heavily on the recognition of a clinical syndrome consistent with the diagnosis in the absence of signs of an alternate cause of thrombotic microangiopathy. Limited understanding of the epidemiology, genetics, and clinical features of aHUS has the potential to delay diagnosis and treatment. To advance our understanding, a more complete characterization of the unique phenotypical features of aHUS is needed. Further studies to identify additional genetic loci for aHUS and more robust biomarkers of both active and quiescent disease are required. Advances in these areas will undoubtedly improve the care of patients with aHUS.
Introduction
Atypical hemolytic uremic syndrome (aHUS) is a disorder belonging to the category of diseases known as thrombotic microangiopathies (TMAs; Figure 1). The pathologic lesion that defines all of the TMAs includes thickening of arterioles and capillary walls, prominent endothelial swelling and detachment, and subendothelial accumulation of proteins and cell debris. The ultimate outcome is fibrin and platelet-rich thrombi obstructing vessel lumina with resultant tissue ischemia. The laboratory correlate of a TMA is a microangiopathic hemolytic anemia that manifests as a mechanical, nonimmune hemolytic anemia; fragmented erythrocytes (schistocytes); and thrombocytopenia. Markers of intravascular hemolysis include an elevated plasma lactate dehydrogenase (LDH) and undetectable or reduced plasma haptoglobin level. An increase in indirect bilirubin, plasma aspartate aminotransferase (AST), and plasma free hemoglobin are seen in severe cases.

The TMAs. Relationship of the thrombotic microangiopathy lesions. (1) TMAs are designated by thrombocytopenia, hemolysis, and schistocytes on peripheral smear. (2) Alternatively, thrombocytopenia, hemolysis, schistocytes, and renal failure define the HUS syndromes. (3) TTP is theoretically excluded after an assessment of the ADAMTS 13 activity.
The TMAs. Relationship of the thrombotic microangiopathy lesions. (1) TMAs are designated by thrombocytopenia, hemolysis, and schistocytes on peripheral smear. (2) Alternatively, thrombocytopenia, hemolysis, schistocytes, and renal failure define the HUS syndromes. (3) TTP is theoretically excluded after an assessment of the ADAMTS 13 activity.
Despite the striking pathological and clinical similarities, the TMAs may be separated into 2 broad categories; thrombotic thrombocytopenia purpuras (TTPs) and HUS. The former was well described by George et al and has been updated in 2 of the accompanying education articles in this issue and will not be addressed further.1,2 HUS is a TMA accompanied by renal impairment. In this setting, the renal microvasculature is the predominant site of TMA; however, other organ systems, particularly the brain, heart, lungs, and gastrointestinal tract, may be affected. HUS is broadly classified as typical or atypical with the acknowledgment that there are very many syndromes that do not fall into the currently accepted view of these forms of HUS. The typical form of HUS, often appropriately referred to as STEC-HUS (for Shiga toxin–producing Escherichia coli HUS) is secondary to Shiga toxin–producing organisms, such as enterotoxigenic E coli (O157:H7 or O104:H4) or Shigella, and begins with a history of bloody diarrhea in the majority of cases.3
The designation of aHUS is best reserved for the TMA that is associated with dysregulation of the alternative pathway of complement.
Other disorders, such as malignant hypertension, septicemia, autoimmune disorders (eg, lupus erythematosus, scleroderma renal crisis, and antiphospholipid Ab syndrome), streptococcal infections in children, and malignancy, can cause secondary HUS syndromes, as can pregnancy and the HELLP (hemolysis, elevated liver enzymes, and low platelets) syndrome. These entities represented here by the designation “secondary” HUS should be considered as part of the disease that presumably triggered the TMA: that is, malignant hypertension-, chemotherapy-, or pregnancy-associated HUS.
Finally, we acknowledge a fourth category of HUS that others have designated “idiopathic” because of the presence of TMA in the absence of a diagnosis of TTP, aHUS, or an accepted secondary disease association. In the past, this term had been used to designate nonsecondary forms, including those with mutations that affect the alternate pathway complement activation.2,4 This group of patients has accounted for nearly 50% of cases in previous cohort reports.4 Now that complement pathway mutations are known to cause aHUS, we suggest that the designation idiopathic HUS should only be used after genetic screening has ruled out aHUS as the more appropriate diagnosis and when an alternate, secondary association cannot be found. As more genetic loci for aHUS are discovered, fewer patients will fall into the idiopathic category.
The AP and aHUS
The alternate complement pathway (AP) is constitutively active and functions as an arm of our innate immune system (Figure 2). Tight control of the AP is required to limit unregulated generation of C3 convertase and subsequent generation of the C5 convertase. C5 convertase activity leads to cleavage of C5 and liberation of C5a, an anaphylatoxin. The sequential assembly of C5b and C6-C9 to form the membrane attack complex5 at the vascular endothelial cell surface causes endothelial cell damage, platelet activation, and thrombus formation.

The AP. The AP begins with activation of C3 and leads to the assembly of the membrane attack complex as a mechanism of protection from infectious agents. Dysregulation of this pathway in disease is most often by the loss of function of regulatory proteins and can lead to endothelial injury, platelet activation, and thrombosis. Regulatory genes (−) that have been shown to be mutated in aHUS are shown in gold. CFH, CFI, MCP, and THBD cooperate to regulate complement activation or inactivate endothelial cell surface-bound C3b, protecting endothelial cells from complement-mediated injury. C3 mutations affect the ability of C3 to bind to regulatory proteins and CFB mutations are gain-of-function mutations (+) that result in an increased stability of the C3 convertase.
The AP. The AP begins with activation of C3 and leads to the assembly of the membrane attack complex as a mechanism of protection from infectious agents. Dysregulation of this pathway in disease is most often by the loss of function of regulatory proteins and can lead to endothelial injury, platelet activation, and thrombosis. Regulatory genes (−) that have been shown to be mutated in aHUS are shown in gold. CFH, CFI, MCP, and THBD cooperate to regulate complement activation or inactivate endothelial cell surface-bound C3b, protecting endothelial cells from complement-mediated injury. C3 mutations affect the ability of C3 to bind to regulatory proteins and CFB mutations are gain-of-function mutations (+) that result in an increased stability of the C3 convertase.
The first indication that excessive activation of the AP was associated with aHUS came in 1973 with the report of 5 patients with HUS and low plasma C3 levels. Since that time, several dysregulated complement pathway proteins have been identified in patients with aHUS, both with and without decreased C3 levels, and it is now known that a decreased C3 is not universal in aHUS.4,6
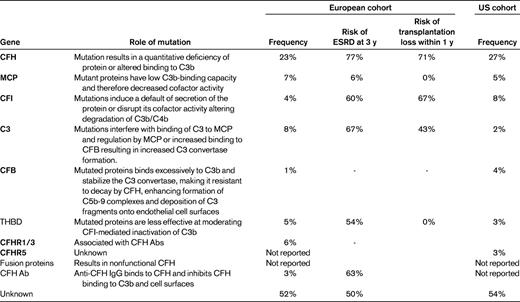
It has been shown previously that mutations in genes encoding proteins that regulate the AP or autoantibodies that inhibit complement regulatory proteins can be identified in approximately 60%-70% of aHUS patients.4,7 Both mutations that result in a quantitative deficiency of protein and mutations associated with a normal plasma level of functionally ineffective protein have been identified. Therefore, measurement of complement factor levels cannot substitute for mutation screening in patients suspected of aHUS.
Complement factor H (CFH) mutations are the most common and account for 23%-27% of identified mutations in registry patients in the United States and Europe4,7 (Table 1). Membrane cofactor protein (MCP) mutations occur at a frequency of 5%-7%. Complement factor I (CFI) and complement component C3 (C3) occur at 4%-8% and 2%-8%, respectively. Gene mutations in complement factor B (CFB; 1%-4%) and thrombomodulin (3%-5%) have also been noted.8,9
The CFH-related (CFHR) proteins have also been associated with aHUS. The role of the CFHR proteins is less well defined than the other complement proteins; however, complement-modulatory activity has been reported.10,11 Homozygous deletions in the CFH-related proteins 1 and 3 make up approximately 6% of cases, with homozygous deletions being associated with CFH autoantibody levels for unclear reasons.4,8,12–16
CFH is in close proximity to the genes CFHR1-CFHR5 encoding the 5 CFHR proteins. The high degree of sequence homology that exists between CFH and the CFHR genes results in deletions and substitutions within the CFH gene through nonallelic homologous recombination. The resulting hybrid proteins are often poorly functioning and may affect the regulatory role of the native CFH protein. These mutations account for 1%-3% of aHUS patients.4 Up to 12% of patients have mutations in 2 or more genes. Finally, 6%-10% of patients, primarily children, have an acquired risk for aHUS via anti-CFH autoantibodies.4,17,18 Many of these patients appear to also have the homozygous CFHR1-CFHR3 deletions; however, they may also be associated with other aHUS mutations.
Despite the genetic advances that have been made in aHUS, 35%-40% of patients with a clinical scenario consistent with aHUS will have no demonstrable genetic mutation using current screening strategies. These patients may have mutations in unscreened regions of known genes. Alternatively, there are likely to be additional genes that cause aHUS that are not yet discovered. To further complicate this scenario, even when genetics are well described in a given family, the penetrance of the disease is only 50%, with half of family members with the same mutation remaining healthy. This may be a consequence of modifier genes or the influence of environmental factors.13
Presentation and diagnosis of aHUS
The data on clinical presentation of aHUS are limited by patient number and the robustness of information ascertained at presentation and come primarily from 2 cohorts.4,12 In the largest cohort of 273 patients, aHUS affected both children and adults, with onset during childhood being only slightly more frequent than in adulthood (Table 2).4,19 The male to female ratio was equal in children; however, there was an increased ratio of female aHUS patients in the adult years.12,13 An infectious event such as an upper respiratory tract infection or gastroenteritis triggered the onset of aHUS in nearly half of the patients.4 Although the association with diarrhea has been well established with STEC-HUS, diarrhea also preceded aHUS in up to 24% of aHUS cases.4 It remains unclear whether the diarrheal episode acted as a trigger or was a result of the TMA. As many as 20% of women with aHUS experienced their first episode around the time of a pregnancy, with the majority of these (80%) being in the postpartum period.4,20,21 In the European cohort, 16% of aHUS cases were reported as a secondary HUS, with malignant hypertension being the leading cause of the secondary forms. The secondary forms of HUS highlight the difficulty in separating aHUS from other causes of TMA. When a secondary HUS occurs, such as in the setting of septicemia, malignant hypertension, chemotherapy, malignancy, or coincident with an autoimmune or rheumatologic disorder, signs and symptoms of the primary disease can confound the diagnosis of aHUS.
Both children and young adults with aHUS have nonspecific symptoms of illness: pallor, poor feeding, vomiting, fatigue, and drowsiness. Anuria or oligoanuria with or without peripheral edema may be present. Marked hypertension may also be present either from the acute kidney injury or from the ischemia caused by the TMA. Hypertension may be severe enough to provoke posterior reversible encephalopathy or cardiac failure. Half of children and the majority of adults need dialysis at admission. Extrarenal manifestations are observed in 20% of patients.4,12 The most frequent extra renal manifestation is CNS involvement (10% of patients) with diverse presentations: irritability, drowsiness, seizures, diplopia, cortical blindness, hemiparesis or hemiplegia, stupor, and coma. Myocardial infarction due to cardiac microangiopathy has been reported in approximately 3% of patients and is presumed to be the cause of reported episodes of sudden death.4,22 Five percent of patients present with a life-threatening multiorgan failure due to diffuse TMA4,12
Less commonly, aHUS patients have more of an insidious onset, with subclinical anemia and fluctuating thrombocytopenia for weeks or months and apparent normal renal function at diagnosis.12 Unusual presentations of aHUS are possible. Some patients have little or no anemia or thrombocytopenia and the only manifestation of an active TMA is hypertension and proteinuria with or without an overtly abnormal creatinine (Figure 3).

A patient with aHUS. The patient is a 45-year-old woman with a longstanding history of untreated hypertension who presented to primary care for foot and ankle swelling and was found to have a blood pressure of 184/102 and a creatinine of 7.2 (636 mmol/L). Her C3 level was one-third of normal, the lactic acid dehydrogenase was elevated, the haptoglobin was decreased, and the platelet count was in the normal range. The initial diagnosis of TTP was entertained. The patient achieved some degree of renal recovery with the start of PE. She received 6 doses of PE peri-presentation, but then discontinued it because of self-reported unacceptable side effects. The patient declined to start eculizumab and was subsequently lost to follow-up. Upon re-presentation 6 months later, she required dialysis and was started on eculizumab. Seven months after re-presentation, the patient remains on dialysis and genetic studies demonstrated a mutation in C3. A corresponding color normal bar has been added to each graph. This case highlights several interesting aspects of aHUS: the platelet count does not have to be below 150 000 K/mm2, the diagnosis of aHUS can be delayed by the consideration of alternate TMA-causing disease; patients may present with late-stage renal disease; plasma exchange can be effective, but may be difficult for patients to sustain; a late start of anticomplement therapy can recover the hematologic findings of aHUS, but may not facilitate renal recovery; genetic testing cannot be part of the acute management; and C3 levels may remain low even when hematologic remission is achieved.
A patient with aHUS. The patient is a 45-year-old woman with a longstanding history of untreated hypertension who presented to primary care for foot and ankle swelling and was found to have a blood pressure of 184/102 and a creatinine of 7.2 (636 mmol/L). Her C3 level was one-third of normal, the lactic acid dehydrogenase was elevated, the haptoglobin was decreased, and the platelet count was in the normal range. The initial diagnosis of TTP was entertained. The patient achieved some degree of renal recovery with the start of PE. She received 6 doses of PE peri-presentation, but then discontinued it because of self-reported unacceptable side effects. The patient declined to start eculizumab and was subsequently lost to follow-up. Upon re-presentation 6 months later, she required dialysis and was started on eculizumab. Seven months after re-presentation, the patient remains on dialysis and genetic studies demonstrated a mutation in C3. A corresponding color normal bar has been added to each graph. This case highlights several interesting aspects of aHUS: the platelet count does not have to be below 150 000 K/mm2, the diagnosis of aHUS can be delayed by the consideration of alternate TMA-causing disease; patients may present with late-stage renal disease; plasma exchange can be effective, but may be difficult for patients to sustain; a late start of anticomplement therapy can recover the hematologic findings of aHUS, but may not facilitate renal recovery; genetic testing cannot be part of the acute management; and C3 levels may remain low even when hematologic remission is achieved.
The variability of presenting symptoms and the inability to rule out other forms of TMA accounts for much of our failure to classify patients as aHUS at presentation. The clinical context, predominant symptoms, and basic laboratory data at presentation may allow differentiation into TTP or typical HUS (Figure 4). The presence of a severe ADAMTS 13 deficiency allows a tentative diagnosis of TTP in some patients. The presence of a classic epidemic of bloody diarrhea or a confirmed positive Shiga toxin test will lead to the diagnosis of STEC-HUS. Not all of the remaining patients will be or should be classified as aHUS. aHUS should be considered when TTP or typical HUS seem less likely: when no other disease can be identified that would account for the signs and symptoms, and particularly when the systemic C3 complement level is low. This distinction is important, because many of the secondary TMAs will respond to treatment of the primary disease and an aHUS treatment algorithm would not need to be activated.

Assessment of HUS. Because there are no specific acute markers that distinguish aHUS from the other TMAs, a screening and treatment strategy for HUS will depend on the clinical presentation and the presumed diagnosis. In the absence of an easily identifiable cause of the TMA, a broader range of laboratory tests will be required to formulate a likely diagnosis. The other laboratory tests to consider when assessing patients with TMA are: ADAMTS13, C3, C4, CFH serology, CFI serology, methylmalonic acid (urine and plasma), homocystine, HIV, ANA, lupus anticoagulant, antiphospholipid antibody, and direct antiglobulin. In addition, very young patients should be evaluated for streptococcal infection as a possible cause of their HUS. Genetic testing should be done early in suspected cases of aHUS; however, treatment should not be delayed pending results.
Assessment of HUS. Because there are no specific acute markers that distinguish aHUS from the other TMAs, a screening and treatment strategy for HUS will depend on the clinical presentation and the presumed diagnosis. In the absence of an easily identifiable cause of the TMA, a broader range of laboratory tests will be required to formulate a likely diagnosis. The other laboratory tests to consider when assessing patients with TMA are: ADAMTS13, C3, C4, CFH serology, CFI serology, methylmalonic acid (urine and plasma), homocystine, HIV, ANA, lupus anticoagulant, antiphospholipid antibody, and direct antiglobulin. In addition, very young patients should be evaluated for streptococcal infection as a possible cause of their HUS. Genetic testing should be done early in suspected cases of aHUS; however, treatment should not be delayed pending results.
Treatment of aHUS
Plasma therapy
Plasma therapy in the form of plasma exchange or plasma infusion has been the cornerstone of aHUS therapy since the 1980s and was essentially the only therapy available until recently. The effectiveness of plasma therapy is presumed to be related to its ability to deliver normal levels of CFH, CFI, CFB, and C3 and, when plasma is exchanged by apheresis, to remove mutant CFH, CFI, CFB, C3 and anti-CFH Abs.20,23–25 Plasma therapy has not been studied in a controlled fashion, so reported successes or failures of therapy should be interpreted cautiously. Registry data suggest that plasma therapy is at least 70% effective in achieving hematologic remission. Renal remission appears to be less certain and may relate to the particular plasma regimen used or the time lapse from disease onset to the start of therapy.4 The determination of whether to perform plasma exchange or infusion is based on several factors. In the ideal setting, it is based on the presumed pathology. Plasma exchange is preferred when an abnormal, possibly competing protein exists and infusion is an acceptable alternative to exchange if a protein deficiency exists. The size of the patient and the degree of renal impairment may limit eligibility for infusion.
The European Pediatric Study Group for HUS has made expert recommendations for the treatment of patients with aHUS that include daily therapy for 5 days and then a graded decrease based on response.23 It has been recommended that plasma therapy be started within 24 hours of presumed diagnosis of aHUS, because this may be the key to effective plasma therapy.26 Because the genetic information and serologic results are likely not to be available at the time of first presentation, plasma exchange will be the preferred treatment in all but those patients in whom patient size or local expertise prohibits exchange. Plasma infusions or exchange should be performed daily until the platelet count, LDH, and hemoglobin levels are substantially improved or even normalized or until an alternate treatment strategy has been decided upon. Renal function is an important marker to follow during the use of plasmatherapy, especially in the setting of acute kidney injury; however, care must be taken to not use the creatinine to guide plasmatherapy when the patient may have irreversible renal injury. When laboratory parameters have improved substantially, the intensity of therapy can be reduced. Persistence of hemolysis or lack of improvement in thrombocytopenia after 3-5 days of plasma therapy should be considered nonresponse to therapy, and is an indication to stop plasma exchange and begin eculizumab. Patients with aHUS from MCP mutations are unlikely to respond to plasma therapy because MCP protein is not a plasma protein. Secondary forms of HUS may also be less responsive to plasma therapy.
Plasma therapy is not without its challenges and, even when it is successful, there may be logistical and technical limits to the ease with which therapy can be continued. Plasma therapy may need to be discontinued if there is lack of vascular access secondary to infection or thrombosis, if anaphylaxis or intolerance to the procedure, or when an experienced infusion/apheresis center.
Eculizumab therapy
The activation of the terminal complement pathway is essential for the development of the endothelial lesion that characterizes aHUS.27,28 Eculizumab (trade name Soliris; Alexion Pharmaceuticals) is a recombinant, humanized, monoclonal Ig that targets C5 and blocks the cleavage of C5 to C5a and C5b, preventing the generation of the proinflammatory peptide C5a and the membrane attack complex C5b-9. Eculizumab, previously used for paroxysmal nocturnal hemoglobinuria was approved both by the European Medicines Agency (EMA) and by the US Food and Drug Administration (FDA) in September of 2011 for the treatment of aHUS after successful trials in adults and adolescents. The recommended weight-based dose set by the manufacturer (available from the Soliris package insert) is initially given weekly as induction therapy and then transitions to every other week.
There are now several case reports supporting its effectiveness in aHUS both before end-stage renal disease (ESRD) and after kidney transplantation.29–31 It is not yet clear how to monitor response to therapy, because the more traditional markers of TMA (ie, platelet count, LDH, and haptoglobin) may not be sensitive enough to determine whether full complement blockade is achieved. Some practices, ours included, use a terminal complement marker (such as a functional C5 level; Mayo Medical Laboratories) to determine whether the terminal complement pathway is adequately blocked (see Nester et al41 and L. Xie, Y. Zhang, C.M.N., A.I. Reed, R.J. Smith, C.P.T., Prophylactic eculizumab in the management of CFH-mediated atypical hemolytic uremic syndrome in an adult kidney transplant recipient, Transplantation Proceedings 2013, in press). Monitoring in this fashion in conjunction with usual markers of TMA and physical signs and symptoms can be used to adjust the dose or the interval between doses to achieve full complement blockade.
The major concern with eculizumab treatment is the risk for infection with encapsulated bacterial organisms, particularly Neisseria meningitis, as a result of terminal complement blockade.32 Therefore, patients must receive meningococcal vaccination before being treated with eculizumab (and covered with appropriate antibiotics for 14 days if there is not enough time to wait for the immune response). However, because no vaccine presently protects against the B serotype, and because patients with chronic kidney disease may have lower rates of seroconversion, patients and physicians must decide if long-term prophylactic antibiotics are required despite vaccination. Ideally, patients should also receive the pneumococcal and Haemophilus influenzae vaccinations before dosing.
With the availability of eculizumab and its relative ease of administration, there is now a choice of plasma therapy or eculizumab for acute therapy. As has been proposed by Loirat et al, plasma exchange would be a reasonable first therapy, with eculizumab introduced for nonresponse to plasma or with transition to the outpatient setting, thus allowing time for successful meningococcal vaccination.3 After the genetic studies are available, a longer-term plan for plasma therapy (ie, continuation with eculizumab or liver transplantation) can then be made based on safety, efficacy, and cost. Barriers to eculizumab use in the United States at this time include physician inexperience with aHUS and/or eculizumab, patient safety concerns, functional lack of immediate access to the drug, and cost.
Liver transplantation
Because CFH, CFI, CFB, and C3 are synthesized in the liver, liver transplantation remains an appropriate option for some patients to provide a source of normal protein. There have been several liver transplantations performed in patients with aHUS.33–40 Although the first 3 transplantation patients died, the subsequent liver transplantations (with and without simultaneous renal transplantation) after appropriate perioperative conditioning protocols have resulted in acceptable outcomes with no reported recurrences of aHUS to date. The recommendation of a liver transplantation requires consideration of the risks and benefits, including the experience of the transplantation center. If a kidney transplantation is required for the ESRD patient, a liver-kidney transplantation should be considered.
Renal transplantation
Several patients with aHUS will progress to ESRD because of inadequate response to therapy or even delay in diagnosis. Other patients will present at ESRD. aHUS patients who need renal transplant require particular attention because of the high rate of recurrent disease without specific therapy in all patients except those with a mutation in MCP. There are no trials to support a required approach for renal transplantation; however, at the minimum, it has been recommended that a complete genetic investigation before transplantation be performed for known genes associated with aHUS, as well as a serologic assessment for relevant autoantibodies.41 Patients with aHUS should undergo a renal transplant under the cover of eculizumab, and 1 or 2 sessions of preoperative plasma therapy should be considered.43 Even in patients in whom MCP is the only documented genetic mutation, another unidentified mutation in a second complement regulatory protein is possible and consideration should be given to using eculizumab, at least in the perioperative setting (especially if an abnormal C3 is present in the patient). In addition to other routine pretransplantation vaccination recommendations, meningococcal vaccine should be given to reduce the infectious risk when eculizumab therapy is used. Special attention must be given to the chronic dialysis patient, who is less likely to respond to immunization and in whom the risk for meningococcal infection may remain high after immunization.
Because current recommendations include an option for pretransplantation aphaeresis and eculizumab, a living donor is preferred. Given the number of aHUS patients who remain without identifiable genetic mutations and those with multiple genetic defects, a genetic screen of a related donor does not eliminate the risk for aHUS in that donor, and living related donation remains controversial. A living unrelated donor is therefore the ideal choice for optimal outcome after a renal transplant alone.
Conclusion
Several issues require attention to maximize the care and treatment of patients with aHUS, not the least of which is determining which patients actually have aHUS. The TMAs may be indistinguishable clinically from each other, making a formal diagnosis difficult. At presentation, it is reasonably easy to recognize typical HUS, and we are becoming more certain about the diagnosis of TTP (defined by ADAMTS13 studies). Gains in the understanding of the genetic mechanism behind aHUS have been very informative in eventually classifying aHUS patients, but genetic tests are not available at presentation and not all patients have an identifiable genetic mutation. The early biomarkers that would help us distinguish the alternative forms of HUS syndromes from aHUS are missing, and the clinician must still rely on history, physical findings, and laboratory parameters at presentation to categorize individual TMAs.
Not only are our current biomarkers limited in their ability to separate the TMAs, they also fail in some cases to help us determine whether the disease is active or in remission on therapy. Because a normal CFH or other regulatory protein level does not exclude the presence of a mutation, even our more acutely available serologic tests can be limited in helping us achieve a diagnosis.
With this as background and given the mechanism of possible aHUS treatments and their risks, benefits, and costs, the challenge is to define the best treatment choice for each individual patient in the absence of genetic confirmation of native disease. In the setting of ESRD in defined aHUS, the transplantation regimen chosen for any given patient must take into account the experience of the treating physicians, access to various treatments, and patient preference considering the short- and long-term risks and benefits of a liver kidney transplant compared with a kidney transplant alone with eculizumab.
What have we learned these past 2 decades? The description of typical HUS and aHUS as diarrhea positive and diarrhea negative, respectively, is no longer tenable given the incidence of diarrheal symptoms in aHUS. Genetic testing is a valuable tool in confirming an aHUS diagnosis and has led to more precise treatment options. Although we are beginning to understand that HUS occurs in children and adults equally, we are still lacking a full description of the range of organ involvement in aHUS. The identification of the remaining genetic risk factors for disease, understanding why the penetrance of aHUS is only 50%, determining whether all TMAs share similar molecular pathophysiology or genetic foundations, and identifying more robust biomarkers will be the most remarkable future advances in understanding this disease. Armed with this information, we will be able to counsel families and patients more thoroughly, define which family members should be excluded from organ donation, and determine whether mothers should be assessed for their risk of TMA before pregnancy. The ultimate goal is to continue to advance our therapeutic regimens to prevent ESRD while improving the daily lives of aHUS patients.
Disclosures
Conflict-of-interest disclosure: C.M.N. is on an advisory committee for Alexion. C.P.T. declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Carla Nester, MD, MSA, Assistant Professor, Director, Pediatric Glomerular Disease Clinic, University of Iowa Hospitals and Clinics, 200 Hawkins Dr, BT 4036, Iowa City, IA 52242-1081; Phone: 319-353-7335; Fax: 319-384-9616; e-mail: carla-nester@uiowa.edu.