Abstract
Outcomes for patients with hemophilia have improved dramatically over the past 50 years. With the increased availability of safe clotting factor concentrates, the primary focus in clinical management is now the prevention of long-term complications, most notably the debilitating hemophilic arthropathy that is associated with severe disease. This article reviews evidence-based approaches for managing both children and adults with hemophilia. Definitive evidence of improved clinical results from primary prophylaxis started in young patients with severe hemophilia A and a minimal bleeding history is presented. Furthermore, recent studies showing benefits for initiating prophylaxis in older adolescents and adults with established joint disease are examined. Inhibitors to factor VIII are the most problematic complication of factor replacement therapy. Patient-specific and treatment-related factors that contribute to the risk of inhibitor formation are discussed and controversies and clinical evidence related to approaches for tolerance induction are reviewed.
Introduction
Hemophilia A and B are X-linked recessive disorders caused by deficiencies in or absence of coagulation factors VIII and IX (FVIII and FIX), respectively. The overall incidence of hemophilia is 1 in 5000 male births, with hemophilia A making up 80% of cases. Clinical bleeding tendency correlates well with the degree of factor deficiency and is the basis for the classification of hemophilia; severe disease is defined as a plasma factor activity level of < 1 IU/dL, moderate disease 1-5 IU/dL, and mild disease > 5 to < 40 IU/dL.1 Patients with severe disease are prone to frequent episodes of spontaneous bleeding that most often affect joints (predominantly the ankles, elbows, and knees) but can occur anywhere, including muscles and other soft tissue sites, the gastrointestinal tract, and the CNS. Recurrent joint bleeding can lead to chronic arthropathy, the major cause for morbidity in hemophilia. Patients with moderate hemophilia typically bleed only in response to some element of trauma, but with the development of tissue damage from recurrent injury, affected joints and muscles can become more prone to bleed with little or no apparent provocation. Patients with mild hemophilia bleed only in response to significant tissue injury induced by trauma or surgery. The primary therapy for hemophilia is coagulation factor replacement, given either episodically on demand for the treatment of acute bleeds or prophylactically to prevent them.1 Currently, there are several virus-inactivated plasma-derived and recombinant FVIII (rFVIII) and FIX concentrates and standard practice is for patients or their families to learn skills enabling home infusions.
Early prophylaxis
The observation that chronic debilitating joint disease was much less likely to develop in patients with moderate hemophilia compared with patients with severe disease led to the development of prophylactic factor infusion regimens in Sweden beginning in 1958. The Swedish experience demonstrated a lower incidence of arthropathy in individuals with severe hemophilia who were started on prophylaxis as young boys compared with historical controls treated with on-demand therapy.2 However, definitive proof of the efficacy of primary prophylaxis, intended to prevent the onset of bleeding induced joint destruction, came from data in 2 randomized controlled trials that evaluated the initiation of prophylactic factor infusions in young boys who had previously experienced no or few joint bleeds.3,4
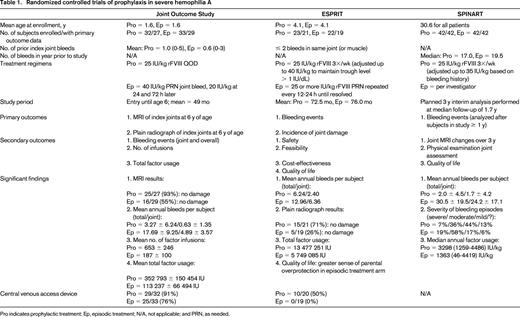
The Joint Outcome Study reported by Manco-Johnson et al randomized 65 boys with a minimal bleeding history (mean of ≤ 1 index joint hemorrhage in each study arm) and < 2.5 years of age at study entry to prophylaxis with 25 IU/kg rFVIII every other day (QOD) or to enhanced episodic treatment, defined as 40 IU/kg rFVIII infused at the time of joint hemorrhage followed by 20 IU/kg doses 24 and 72 hours later. Final evaluation of study subjects at 6 years of age demonstrated that 93% of patients in the prophylaxis arm had no joint damage, as determined by MRI, compared with 55% of patients on episodic treatment.3 Furthermore, significantly fewer joint and overall bleeding episodes were experienced by boys randomized to receive prophylaxis.3 The Evaluation Study on Prophylaxis: a Randomized Italian Trial (ESPRIT) study reported by Gringeri et al evaluated the institution of prophylaxis in a somewhat older cohort of boys (median age 48-50 months) who had no evidence of arthropathy by radiograph or physical examination at randomization. An evaluation of 40 boys randomized to a regimen of prophylaxis (25 IU/kg rFVIII 3 times per week with adjustments up to 40 IU/kg to maintain trough levels > 1% or because of frequent bleeding) or on-demand treatment was performed after a median of 82 months on study.4 Fewer bleeding episodes (joint and overall bleeding) and less radiographic evidence of joint damage was seen in the prophylaxis group, with the best outcomes noted in patients who started prophylaxis before 3 years of age. Furthermore, there was a significant difference in quality of life measures between the 2 treatment arms because of a sense of markedly greater parental overprotection in boys randomized to episodic treatment.4 An overview of both studies is presented in Table 1.
The clear benefits of 3 to 4 times per week factor infusions in primary prophylaxis are offset to an extent by markedly increased costs from more factor consumption and complications from indwelling central venous access devices needed to support intensive treatment regimens in most very young patients.5,6 An alternate approach now under study in Canada is the initiation of primary prophylaxis with a single weekly infusion of 50 IU/kg rFVIII and subsequent stepwise dose escalation if bleeding symptoms are not adequately controlled, first to 30 IU/kg twice weekly and then to 25 IU/kg QOD.7 In an interim report of 25 patients published by Feldman et al, at 5 years into prophylaxis, 40% of subjects still required only once weekly factor infusions and only 28% had escalated to QOD treatment.7 On average, subjects experienced twice as many hemarthroses (1.2 per year) and developed more target joints (defined in the Canadian study as ≥ 3 clinically determined bleeds into any one joint over a consecutive 3-month period) than participants in the prophylaxis arm of the concurrently run United States–based Joint Outcome Study.3,7,8 At the 5-year interim analysis, the Canadian subjects all had normal joints by physical examination and little evidence of arthropathy by plain radiographs. Furthermore, only 10/25 patients on the Canadian dose escalation regimen required central indwelling venous catheters, compared with 29/32 subjects in the prophylaxis arm of the Joint Outcome Study,3,7 and the estimated cost for the first 5 years of treatment was approximately 20% less than standard high-dose prophylaxis.8 However, subsequent MRI studies did show evidence of osteochondral changes in target joints in 50% of the Canadian subjects. Because these examinations were in boys who were on average 2 years older than subjects studied by MRI in the Joint Outcome Study, a direct comparison of MRI results between the 2 studies is problematic.9
A single universal optimal approach for primary prophylaxis is unlikely to be identified due to inherent limitations in studying a rare disease, significant national differences in economic resources available for hemophilia treatment, and individual patient/family preferences. However, important principles that should be stressed in treatment are that early institution of prophylactic factor replacement (ideally by 2-3 years of age) produces better joint outcomes2,4,10 and that compliance is a critical variable for reducing the incidence of breakthrough bleeding.11,12 Factor usage and the associated cost are important variables to consider in prophylaxis. Therefore, alternate approaches for factor dosing, such as the Canadian escalation scheme, should be considered. To that end, the standard approach in the Netherlands is also a model to study. The Dutch have achieved good long-term joint outcomes, as evaluated in older adolescents and young adults, with 2 to 3 times per week lower dose regimens that consume approximately half as much clotting factor concentrate as the standard high-dose approaches used in Sweden and studied in the Joint Outcome Study and ESPRIT.13 Finally, although there are few data comparing outcomes of on-demand versus prophylactic treatment approaches in patients with hemophilia B, the same rationale for dosing to maintain FIX trough activity levels > 1% has been applied by many treatment centers.14,15
Prophylaxis in adults
Approximately one-third of patients who start primary prophylaxis choose to switch to on-demand therapy as young adults.16 Approximately one-third who make the switch then revert back to prophylaxis.16 As reported in a cohort of Danish and Dutch patients with severe hemophilia in their mid-20s, after a median follow-up of 3.6 years from permanent discontinuation of prophylaxis, more joint bleeds and lower factor consumption rates were seen in patients who switched to episodic treatment compared with those who remained on prophylaxis.17 However, bleeding rates were still quite low in patients who switched, with a median of only 3.2 joint bleeds per year, far less than what has been reported in most cohorts of adults with severe hemophilia on episodic treatment.12,17-19 Furthermore, joint disease, as measured by physical examination and plain radiographs, was similar in the 2 groups of patients.17 Because this was an observational study with short-term follow-up on a self-selected population, no definitive conclusions about the long-term outcomes of switching from prophylaxis to on-demand treatment can be reached.
There is a growing body of evidence showing benefits to instituting prophylaxis regimens in older adolescents and adults with severe hemophilia. The first study to support initiating prophylaxis in patients with established joint disease was a 6-year international longitudinal orthopedic outcome study showing less progression of established arthropathies, as measured by serial physical examinations and plain radiographs, in patients treated with full-time factor prophylaxis regimens.20 More recently, Collins et al used a crossover study design to compare outcomes from 6 months of on-demand treatment with 6 months of a prophylaxis regimen (20-40 IU rFVIII 3 times weekly) in 20 adults 30 to 45 years of age. During on-demand treatment, subjects experienced a median of 15.0 joint bleeds compared with a median of 0 bleeds of any kind on prophylaxis. However, other than a decrease in bleeding rates, this study did not show an improvement from prophylaxis in joint function outcomes or in quality of life measures.18 Another recent study evaluated 66 subjects (median age 26 years) maintained on 6 months of on-demand treatment and then randomized to a year of standard QOD (20-40 IU/kg/dose) or pharmacokinetic-based every third day (20-80 IU/kg/dose) prophylactic infusions of rFVIII. The median annualized bleeding rate for all subjects on prophylaxis was reduced by 99% compared with on-demand treatment, with equivalent results in both prophylaxis arms. Furthermore, from the start to the end of prophylaxis treatment, health-related quality of life measures related to pain and physical functioning showed significant improvements.12
The ongoing international Trial to Evaluate the Effect of Secondary Prophylaxis With rFVIII Therapy in Severe Hemophilia A Adult Subjects Compared to That of Episodic Treatment (SPINART) clinical trial is the first randomized controlled study to evaluate outcomes in adults (mean age 30.6 years) with severe hemophilia A treated with standard prophylaxis (3 times weekly dosing of 25 IU/kg rFVIII with escalation up to 35 IU/kg/dose for poor control of bleeding) or on-demand therapy.19 An interim report of 84 subjects at a median follow-up time of 1.7 years showed a 93% decrease in the frequency of bleeding episodes in patients randomized to the prophylaxis arm. Moreover, as self-reported by study subjects, bleeding events occurring on prophylaxis were significantly less severe.19 SPINART is designed as a 3-year study and final results will include evaluations of index joints by MRI and physical examination and health-related quality of life measures. An overview of SPINART is presented in Table 1.
Final results from SPINART and the Italian prospective 5-year observational Prophylaxis versus On-demand Therapy Through Economic Report (POTTER) study21 will help provide a better understanding of the benefits and costs of instituting prophylaxis in adults with severe hemophilia A and established joint disease. In addition to joint health, another compelling reason for using lifelong prophylaxis in severe hemophilia A is the associated 50% reduction in the risk of intracranial hemorrhage found in inhibitor negative patients.22 Moreover, because the average age at death from intracranial hemorrhage was in the 40s, discontinuation of primary prophylaxis in adulthood could not be expected to mitigate this risk. One of the major barriers of instituting and/or maintaining prophylaxis regimens in adults is the associated cost. Therefore, strategies to achieve the most cost-effective outcomes must be sought. It may be possible to realize good control of bleeding symptoms in adults using considerably less factor concentrate than what has been used in most studies to date. Dosing and infusion schemes in adult studies were extrapolated from the pediatric experience, but the half-life of FVIII increases significantly with age.23 As a result, 48-hour trough FVIII levels in the range of 3-6 IU/dL were seen with standard prophylaxis dosing in recent studies on adult patients.12,18 Trough levels of > 1% could be maintained in most patients with one-third to one-half as much factor as was used in these adult prophylaxis studies if a 48-hour dosing interval was maintained. Because bleeding patterns are influenced by several factors, including poorly understood inherent differences in coagulability, underlying joint damage, and patient activity levels, pharmacokinetic-based dosing can only be considered as a starting point. Dosing refinements need to be based on observed bleeding rates and further tailored to an individual's schedule of activities.24
Mild and moderate hemophilia
As expected by the range of baseline factor activity levels meeting criteria for moderate hemophilia, patients with this diagnosis demonstrate a very wide range of clinical severity. However, bleeding symptoms can be significant and overall approximately 40% of patients with moderate hemophilia report having experienced a joint bleed in the previous 6 to 12 months.25,26 Furthermore, in 20% to 30% of patients, prophylactic factor infusions are used for at least some period of time to manage symptoms.25-27 Retrospective data show that a baseline factor activity level of ≤ 2 IU/dL combined with a history of a first joint bleed before the age of 5 years predicts a bleeding phenotype that is similar to severe hemophilia.27 For these individuals, early institution of prophylaxis should be considered to prevent the development of disabling arthropathies.27
For the majority of patients with mild and moderate hemophilia, the focus of therapy is to provide adequate hemostasis to treat trauma-related bleeding and to prevent bleeding with surgery or other invasive procedures. In addition to infusing factor concentrate, therapeutic levels of FVIII can also be achieved in many patients by intravenous, subcutaneous, or intranasal administration of desmopressin (1-deamino-8-D-arginine vasopressin [DDAVP]).28,29 The main advantages of DDAVP are reduced cost and ease of subcutaneous or intranasal delivery. Responses to clinically relevant levels (> 30 IU/dL plasma FVIII activity) are seen in 66% to > 90% of treated patients, with most responders achieving levels > 50 IU/dL.28,30-32 DDAVP does not raise FIX levels and some specific hemophilia A genotypes are associated with a consistently poor peak FVIII response or short response duration.30-32 However, even in individuals with favorable genotypes, poor responses are frequent, so it is critical to perform a test infusion before therapeutic use. Furthermore, there is an association between age and DDAVP responsiveness, so individuals who respond poorly at a young age should be retested as adolescents or adults.28,30 Because of tachyphylaxis and concerns about hyponatremia leading to seizures and even cerebral edema, DDAVP should not be dosed for more than a few days consecutively and water intake should be limited.28,33 Furthermore, DDAVP should not be used in patients with known or suspected atherosclerosis based on multiple case reports of arterial thrombotic events after its infusion.28
In hemophilia of any severity, the antifibrinolytic agents tranexamic acid and epsilon aminocaproic acid can be used as hemostatic adjuvants to factor replacement therapy or DDAVP to improve clot stability.33,34 Antifibrinolytics are particularly useful for controlling bleeding after dental extractions because they counteract the significant endogenous fibrinolytic activity found in the oral cavity. They can be given topically and/or systemically and typically for a week to 10 days after dental extractions.1 Because of a high incidence of renal colic and ureter obstruction in hemophilia patients treated for hematuria with aminocaproic acid, the use of antifibrinolytics is contraindicated in this setting.35
Inhibitors
Alloimmune inhibitory antibodies to FVIII (referred to as “inhibitors”) develop in 25% to 30% of patients with severe hemophilia A and currently represent the most significant complication to factor replacement therapy.36-38 FVIII inhibitors typically develop in young children, with initial detection after a median of 14 to 16 treatment days and rarely after 50 treatment days.39-41 Inhibitors to FIX in hemophilia B are much less common, occurring in only ∼ 3% of patients, but are often associated with an anaphylaxis type reaction and the development of nephrotic syndrome, both of which substantially complicate attempts at tolerization.42
In hemophilia A, the F8 gene mutation is an important risk factor for inhibitor development, with null mutations (yielding no measurable circulating protein) conferring a significantly higher risk than missense mutations.43 Large gene deletions that span multiple exons are associated with the highest risk of inhibitor formation and similar inhibitor development rates have been reported for most other null genotypes, including nonsense mutations and intron 22 and intron 1 inversions.43 Race is another important risk factor, with multiple reports showing approximately twice the incidence of inhibitors in black patients compared with white patients.38 The hemophilia genotype does not appear to explain racial differences in inhibitor formation.38 Viel et al reported an association between the rate of inhibitor formation and the background F8 haplotype in black hemophilia A patients, leading them to speculate that the mismatch between the background haplotype and the FVIII proteins used in standard recombinant concentrates influenced inhibitor risk.44 However, their observation has not been supported by more recent studies.38,45 Other studies have associated a greater inhibitor risk with specific HLA class II alleles and polymorphisms in multiple immunoregulatory genes have been associated with either a greater risk or a protective effect.46-48 However, results across different studies have not been consistent, leaving unresolved the true contribution of genetic risk factors other than the F8 mutation.46-48
Multiple studies show an association between early intensive exposure to FVIII concentrate and increased risk for inhibitor development.39-41,49 Some studies indicate a protective effect of prophylaxis regimens, but this benefit may be most relevant to patients with low-risk F8 mutations.39,40 The relative immunogenicity of rFVIII products compared with plasma-derived products containing high amounts of VWF has been the source of much debate.50 The recently published report by the RODIN group of prospectively collected data in 574 previously untreated patients with severe hemophilia A did not show recombinant factors as a class to be more immunogenic than plasma-derived concentrates and the VWF content of the plasma concentrates used did not appear to influence inhibitor risk.36 However, a subgroup analysis of specific products showed a small but significantly increased relative risk (adjusted hazard ratio of 1.6) for inhibitor formation in patients treated with a second-generation full-length rFVIII concentrate.36 Although the RODIN report represents the most comprehensive attempt to date to answer the question of product class immunogenicity, inherent limitations in the observational nature of the study leave its conclusions open to question.50 Additional data from other studies, such as the ongoing Survey of Inhibitors in Plasma-Product Exposed Toddlers (SIPPET) trial, which randomizes previously untreated patients to treatment with either recombinant or VWF-rich plasma-derived concentrates, may help to resolve the immunogenicity issue more definitively.51
Immune tolerance induction (ITI) by frequent dosing of FVIII concentrates over many months to 3 years is the only proven method for durable eradication of inhibitors. Overall, ITI works 60% to 80% of the time.52 Because almost all published reports of ITI outcomes are from uncontrolled studies that contained few patients, there is no consensus optimal treatment regimen. Retrospective data show better outcomes overall with ITI in patients with a peak historical inhibitor titer < 200 Bethesda units (BU), immediate pre-ITI inhibitor titer < 10 BU, younger age, and presence of an inhibitor for < 5 years.52 F8 mutations associated with a lower risk for inhibitor formation are also associated with higher ITI response rates.53 The only randomized trial to evaluate outcomes in ITI compared results in good-risk patients (although F8 genotype was not considered) who received either high-dose FVIII infusions of 200 IU/kg/d or a low-dose regimen of 50 IU/kg given 3 times weekly.54 This study was stopped early for futility and safety considerations. Although no difference was seen in the percentage of patients who were tolerized in each arm, there were significantly more bleeding complications in the low-dose arm. Most of these bleeding episodes occurred in the first phase (start of ITI to negative titer) of treatment.54 Using a similar rationale as SIPPET, the Study of First Time Immunotolerance Induction in Severe Hemophilia A Patients With Inhibitor at High Risk of Failure: Comparison With FVIII Concentrates With or Without Von Willebrand Factor (RESIST Naive) study was designed as a randomized trial to determine whether ITI outcomes in poor-risk patients were influenced by the VWF content of the concentrates used for tolerance induction.55 Unfortunately, because of poor accrual, RESIST Naive was closed recently (E. P. Ewing, City of Hope, Duarte, CA, personal communication, 2013).
Achieving good hemostasis for the treatment or prevention of bleeding is another important area in the management of inhibitor patients. Patients with high-titer inhibitors (> 5-10 BU) or those who demonstrate an anamnestic response to FVIII need to be managed with bypass clotting factors: either FEIBA, an activated prothrombin complex concentrate, or rVIIa. Neither agent is clearly superior and individual patients may respond better to one product for unclear reasons.56,57 In 2 recent prospective randomized trials in inhibitor patients, prophylactic infusions of FEIBA or rVIIa resulted in significantly reduced bleeding rates compared with management with on-demand episodic therapy.58,59 However, both studies only evaluated outcomes for a 6-month observation period, so the long-term efficacy of this approach and whether it can ultimately improve joint outcomes is unknown.
Future directions
Multiple FVIII and FIX molecules with prolonged in vivo half-lives are currently in clinical trials. Development of these new products is reviewed by Drs Amy Shapiro and Margaret Ragni in other chapters in this publication.60,61 A recent gene therapy trial using a serotype-8 pseudotyped adeno-associated virus vector (AAV8) expressing a self-complementary codon-optimized FIX transgene in patients with severe hemophilia B is the first human study to show durable production of therapeutic amounts of a clotting factor protein.62 The first 10 patients treated in the study all show stable expression of FIX, with plasma activity levels of 1% to 6%, over a follow-up period of 8 to 40 months after treatment. The highest FIX expression levels were seen in the cohort treated with the highest vector dose. Although some subjects in this treatment group did show evidence of increased levels of liver transaminases that were temporally associated with a T-cell response against the AAV8 capsid, the immune responses were controlled with a short course of oral corticosteroids60 (A. C. Nathwani, University College London Cancer Institute, personal communication).
Disclosures
Conflict-of-interest disclosure: The author has received research funding from Bayer and Biogen Idec and has consulted for Biogen Idec. Off-label drug use: None disclosed.
Correspondence
Neil Josephson, Director, Hemophilia Care Program, Puget Sound Blood Center, 921 Terry Avenue, Seattle, WA 98104; Phone: 206-292-6570; Fax: 206-292-4094; e-mail: njoseph@uw.edu.