Abstract
Immune thrombocytopenia (ITP) is an autoimmune-mediated condition that results from antibody-mediated destruction of platelets and impaired megakaryocyte platelet production. ITP patients exhibit severe thrombocytopenia and are at risk for significant hemorrhage. Few randomized trials exist to guide management of patients with ITP. Ultimately, each patient requires an individualized treatment plan that takes into consideration the platelet count, bleeding symptoms, health-related quality of life, and medication side effects. This article provides an up-to-date review of management strategies drawing on links between the expanding amounts of clinical trial data and associated biology studies to enhance understanding of the disease heterogeneity with regard to the complex pathogenesis and response to treatment.
Introduction
The hallmark of immune thrombocytopenia (ITP) is autoimmune destruction of platelets in addition to suppression of platelet production by the bone marrow (BM) megakaryocytes. The diagnosis of ITP depends on demonstration of a platelet count < 100 × 109/L and may be found in isolation (primary) or alongside other autoimmune and medical conditions (secondary).1
ITP can be further classified by disease duration based on the following definitions: newly diagnosed (diagnosis to 3 months), persistent (3-12 months), and chronic (> 12 months).1 Patients who have failed splenectomy are considered to have refractory disease and it has been argued that disease severity should be based on clinically relevant bleeding regardless of the platelet count.1 This review focuses on the current diagnosis and management of ITP, highlighting new advances regarding the pathogenesis of ITP and how these findings translate to treatment strategies.
Clinical presentation and diagnosis
Patients with ITP present with hemorrhage secondary to severe thrombocytopenia. The bleeding manifestations of ITP are highly heterogeneous; however, patients usually experience mild mucocutaneous hemorrhage (severe hemorrhage is uncommon when the platelet count is > 30 × 109/L). Prospective data show that the risk of severe hemorrhage is extremely low for children with ITP regardless of the platelet count.2 In adults, additional modifiers such as existing comorbidities, age, activities, and medications may affect the risk of significant bleeding. To date, there are no predictors for the development of more severe hemorrhage in patients with no or little bleeding at diagnosis.3
The diagnosis of ITP is one of exclusion and antiplatelet antibody testing is not recommended because of high inter-laboratory variability and poor sensitivity.4 Essential components required for making the diagnosis include: personal history, family history evaluating for inherited thrombocytopenias, physical examination, complete blood count with differential, reticulocyte count, and review of the peripheral blood smear.5,6 Patients with ITP demonstrate isolated thrombocytopenia and no additional abnormalities with the exception of anemia in the setting of bleeding. Additional testing for HIV and hepatitis C is recommended for all adult patients with ITP5,6 because both can be associated with ITP and treatment depends upon management of the underlying condition. BM evaluation in patients with no additional findings is of low yield7 and guidelines propose that it should be reserved for patients with atypical features.5,6
More extensive testing should individualize risk, patient symptoms, cost, and availability of testing, as well as the specificity and sensitivity of individual tests. For example, although Helicobacter pylori has been linked to ITP, the data suggest that routine screening of all patients is not a reasonable approach; only a select group of patients with symptoms or those from highly endemic regions should be considered for testing.5
Pathogenesis
ITP is a complex disorder of immune dysregulation; however, the final pathway is loss of tolerance of the immune system to self-antigens located on the surface of the platelets and megakaryocytes (Figure 1).8,9 Table 1 outlines the different roles that both T and B cells have been speculated to play in the development of ITP. Fundamentally, ITP results from antiplatelet antibodies produced by B cells, often targeting primary platelet glycoproteins such as GP IIb/IIIa.9 Beyond the effects on circulating platelets, these antibodies are also directed against platelet glycoproteins on the surface of megakaryocytes, inducing apoptosis-like programmed cell death and reducing platelet production.10,11

Proposed mechanism of immune dysregulation in ITP. (A) T cells are activated upon recognition of platelet-specific antigens on the APCs and therefore induce antigen-specific expansion of B cells. The B cells in turn produce autoantibodies with specificity for glycoproteins expressed on platelets and megakaryocytes. (B) Circulating platelets bound by autoantibody are removed by Fc receptors predominantly by splenic macrophages. (C) Autoantibodies also reduce the capacity of megakaryocytes to produce platelets. Adapted with permission from Wei and Jackson.8
Proposed mechanism of immune dysregulation in ITP. (A) T cells are activated upon recognition of platelet-specific antigens on the APCs and therefore induce antigen-specific expansion of B cells. The B cells in turn produce autoantibodies with specificity for glycoproteins expressed on platelets and megakaryocytes. (B) Circulating platelets bound by autoantibody are removed by Fc receptors predominantly by splenic macrophages. (C) Autoantibodies also reduce the capacity of megakaryocytes to produce platelets. Adapted with permission from Wei and Jackson.8
A proposed model for ITP involves antigen-presenting cells (APCs), which serve the primary function of internalizing and breaking down antigenic proteins into smaller peptides. These peptides are then presented to T cells and, through signaling events, the T cell becomes activated.9,12 Perhaps in certain settings such as inflammation, APCs create cryptic epitopes that are capable of escaping negative selection.9
Following activation, T cells have also been shown to demonstrate alterations in patients with ITP. Early studies indicated that patients with ITP had autoreactive T cells that secreted IL-2 upon stimulation with autologous platelets in an uncontrolled manner.12 Furthermore, the T cells observed are primarily against cryptic rather than native epitopes,13 supporting a role for APCs as critical cells in the development of ITP. In addition, patients with ITP demonstrate an increased Th1/Th2 ratio favoring autoreactive B-cell development.14 Emerging data also support a role for Th17, a novel Th cell, in the development of ITP. Th17 cells produce cytokines such as IL-17 that may further drive the imbalance between Th1 and Th2 cells, therefore favoring autoimmunity.12 Lastly, T-regulatory cells (Tregs) are reduced and impaired in ITP.9,12,15 These cells are important in maintaining self-tolerance by reducing cell-mediated and antibody-mediated immune responses.
As outlined above, the pathophysiology of ITP is complex and many interactions remain undetermined. Increased knowledge regarding the cells and cytokines involved in the development of ITP will lead to the discovery of novel therapeutic options.
First-line therapy for ITP
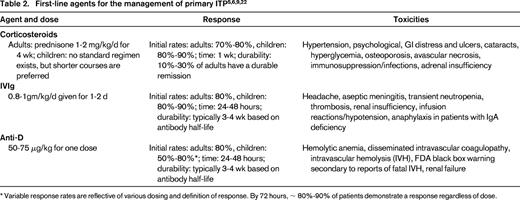
Initial management of ITP is dependent upon factors such as platelet count, patient age, bleeding symptoms, health-related quality of life (HRQoL), need for upcoming procedures, and side effects associated with therapy. Traditional first-line agents include corticosteroids, IVIg, and anti-D immunoglobulin (anti-D). Table 2 outlines the dose, anticipated response rate, and side effects of first-line agents.
Evolving data may change the landscape of first-line therapy with the goal of reducing the number of patients who develop chronic ITP with more aggressive therapy. For example, IVIg influences humoral and cellular immunity by interacting with regulation of Fc receptor expression.16 Furthermore, IVIg effects maturation arrest of dendritic cells (DCs), decreases IL-12 production, and increases the production of IL-10.16 Evidence from a murine model suggests that DCs exposed to IVIg, washed, and then infused can ameliorate ITP. This provides a novel model in which patients' own DCs could be collected, exposed to very small amounts of IVIg, washed, and reinfused.16 These downstream immune changes provide a rationale for the observation that children receiving IVIg at diagnosis were less likely to develop chronic ITP,17 suggesting that IVIg has a long-term immunomodulatory effect. Further studies are needed to confirm this finding and to determine the true benefit of this approach given that the majority of children do not require treatment upfront and the development of chronic ITP in children is uncommon.
The addition of rituximab has been evaluated in adults to provide intensification of upfront therapy. In a recent randomized trial, 133 adults with newly diagnosed ITP were treated with either dexamethasone (40 mg/dose/d × 4 days) alone or in combination with rituximab (375 mg/m2/wk for 4 weeks).18 The primary end point, sustained platelet count > 50 × 109/L at 6 months, was achieved in 58% of the group receiving rituximab and in 37% of the control group (P = .02).18 Results are similar to earlier findings in which patients who received combination therapy with dexamethasone and rituximab had response rates of 63% compared with 36% in patients receiving dexamethasone monotherapy (P = .004).19 One additional study has explored the use of low-dose rituximab (100 mg/dose × 4 doses) in combination with dexamethasone with a 6-month sustained remission rate of 76.2%.20 Lastly, the use of rituximab alone and not in combination with dexamethasone has been explored for nonsplenectomized adult patients with newly diagnosed and relapsed ITP. In this randomized pilot trial, there was no difference in treatment failures (defined by the composite end point of any platelet count < 50 × 109/L, significant bleeding, and need for rescue therapy) between the placebo (21 of 26, 80.8%) and rituximab (21 of 32, 65.6%) groups.21 The investigators highlight that these results suggest that the efficacy of rituximab may not be as profound as previously reported in this patient population and that greater evidence is needed to determine the role of rituximab in this setting.21 Further studies are therefore necessary to understand how rituximab should be incorporated in clinical practice and if escalation of initial therapy with more aggressive immunotherapy is warranted.
Special considerations for children
The majority of children with newly diagnosed ITP and minimal bleeding can be treated with observation alone regardless of platelet count because severe bleeding events are thought be rare.5,6 Should treatment be desired, then initial management can be provided with a short course of corticosteroids, IVIg, or anti-D. If a rapid increase in platelet count is desired, then IVIg and anti-D are preferred based on the ability of these agents to increase the platelet count within 24-48 hours in the majority of children.
Special considerations for adults
Treatment in adults with prednisone is reserved for patients with significant thrombocytopenia (platelet count < 30 × 109/L).22 Data from nonrandomized trials suggest that pulse doses of dexamethasone can induce sustained remission rates in ∼ 50% to 75% of patients23,24 ; however, randomized trials are needed to establish the role of dexamethasone rather than prednisone as initial therapy. IVIg and anti-D are reserved for patients who have a contraindication to corticosteroids or require a more rapid response in platelet count.5,6,22
Second-line therapy for ITP
The most widely studied treatment modalities include splenectomy, rituximab, and the thrombopoietin-receptor agonists (TPO-RAs). Current ASH recommendations for second-line treatment are outlined in Table 3.5 Ultimately, there is a paucity of randomized trials to help guide second-line therapy, so it is critical that individualized treatment plans take into account the patients' perspective, anticipated response rates, and side effects.
Splenectomy
The primary site of circulating platelet destruction is antibody recognition by the Fc receptor on macrophages within reticuloendothelial system. Therefore, splenectomy removes the mechanism of platelet destruction along with a large source of antiplatelet antibody production.
Splenectomy has been the historical second-line therapy for both adults and children with ITP unresponsive to first-line agents and is considered the only “curative” therapy. A systematic review representing 1223 laparoscopic splenectomies showed an immediate response rate of approximately 92%, with 72% of patients having a durable remission at 5 years.25 Results from a large systematic review detailing outcomes of 2623 adults undergoing splenectomy found a composite response rate, defined as a normal platelet count for the duration of follow-up (1-153 months) without additional treatment, of 66%.26 Similar rates are seen in children, with 80% demonstrating a durable remission at 4 years.27 Despite splenectomy having a high success rate, the increasing number of available therapeutic options has caused physicians to be reluctant to universally recommend splenectomy for patients with chronic ITP28 and patients are often hesitant to accept splenectomy as therapy for their ITP.29
The primary side effect of splenectomy is loss of immune protection against encapsulated organisms, which is associated with overwhelming sepsis and infection (hazard ratio of 4.6 between 91 and 365 days and 2.5 beyond 365 days)30 ; however, this risk is likely to be reduced with appropriate presplenectomy vaccinations and postsplenectomy prophylactic antibiotic practices.31 Additional concerns are growing over the possible vascular complications after splenectomy, including the incidence of pulmonary hypertension and venous and arterial thromboembolism.32
Rituximab
Rituximab, a monoclonal CD-20 antibody, was first used in the treatment of clonal B-cell malignancies such as lymphoma. Recognition that rituximab caused rapid depletion of CD-20 positive B cells responsible for antibody production led to its application in autoimmune conditions, including ITP.
Rituximab has been successful in inducing remission in adults33,34 and pediatric patients34,35 with chronic ITP. Two systematic reviews of the adult literature have been published with pooled overall response rates, defined as a platelet count > 50 × 109/L, of 57% (N = 376)34 and 63% (N = 313).33 In addition, 2 systematic reviews have evaluated initial response in children with reported pooled response rates of 57% (N = 116)34 and 68% (N = 323).35 Projected 1- and 5-year response rates, however, were significantly lower at 33% and 26%, respectively, for children and 38% and 21%, respectively, for adults. Predictors of response to rituximab include shorter duration of ITP, secondary ITP, and previous response to corticosteroids; however, these have not been consistently identified across studies.33-35
Findings from clinical trials have resulted in several questions regarding the mechanism of action of rituximab. For example, the delay between B-cell depletion and platelet count response in the majority of patients and why a significant proportion of patients fail to respond at all. In addition, long-term rates of sustained immune tolerance remain low. It remains unclear what accounts for such vast interpatient variability in response. Of particular interest is the finding that rituximab response is associated with changes to the T-cell compartment, such as restoring the Th1/Th2 ratio36 and increasing the number and function of Tregs37 in responders, indicating that perhaps the primary therapeutic effect of rituximab is not simply via B-cell depletion. In addition, in nonresponders, antiplatelet-specific plasma cells have been found to persist in the spleen months after rituximab treatment, presumably accounting for the lack of response to rituximab.38
Side effects of rituximab include infusion-related reactions, infectious complications, and serum sickness. Pooled data from adult studies showed that ∼ 22% of patients experienced a mild or moderate adverse event, with the majority (83%) being related to the infusion.33 There were an additional 10 patients (3.7%) who developed severe or life-threatening events and 9 (2.9%) patients died, 4 from fatal infections.33 Pooled pediatric data revealed that 91% of the adverse events were listed as mild to moderate and were related to the infusion. No pediatric deaths with rituximab have been reported. Additional serious adverse events included infection (N = 3), serum sickness (N = 7), hypersensitivity reaction (N = 2), and the development of common variable immunodeficiency in one child.35 Viral reactivation has also been noted in patients receiving rituximab. Hepatitis B39 reactivation after rituximab commonly occurs and JC virus leading to progressive multifocal leukoencephalopathy has been reported rarely.40
TPO-RAs
TPO-RAs cause stimulation of platelet production by the BM megakaryocyte, leading to an increase in the circulating platelet count. Given that these are not primary immunomodulatory agents, they are not considered “curative” and patients may experience rebound thrombocytopenia upon abrupt drug discontinuation. Two agents, romiplostim and eltrombopag, are currently Food and Drug Administration (FDA) approved for adults with chronic ITP and each acts on different regions of the thrombopoietin receptor. Recent case reports suggest that there is no cross-resistance between the two agents.41,42
Two randomized trials with romiplostim enrolling a total of 125 patients, 63 splenectomized and 62 nonsplenectomized, with a platelet count < 30 [times 109/L43 were published collectively. A durable platelet response, defined as platelet count ≥ 50 × 109/L during 6 or more of the last 8 weeks of treatment, was seen in 38% of the splenectomized patients and in 61% of the nonsplenectomized patients given romiplostim. These results were striking given that only one patient in the placebo group achieved the primary end point.43 In a more recent open label study (N = 234 adults) use of romiplostim resulted in greater platelet response rates (P < .001), lower treatment failures (P < .001), and reduced splenectomy rates (P < .001) compared with standard of care.44
Eltrombopag has similar efficacy to romiplostim, with 2 studies demonstrating that 59% to 81% of patients receiving the drug (−75 mg/d) achieved a platelet count of ≥ 50 × 109/L on day 43 compared with 11% to 16% of the placebo group.45,46 Furthermore, results from the 6-month randomized RAISE study demonstrated that patients treated with eltrombopag had higher platelet count response rates (P < .001), greater reduction in concomitant medications (P = .16), and reduced need for rescue medications (P = .001) compared with patients receiving placebo.47
Long-term figures on both agents suggest that the response in platelet count can be maintained. Data on 200 patients receiving long-term romiplostim therapy (mean duration: 110 weeks) revealed that patients were able to maintain a platelet count of ≥ 50 × 109/L for the majority of the study visits (median: 92% of study visits).48 The EXTEND study, reporting on 299 patients followed for up to 3 years on eltrombopag, demonstrated that 62% of patients had a platelet count ≥ 50 × 109/L without new or increased ITP treatments in > 50% of the visits.49 Beyond a measurable increase in the platelet count, TPO-RAs have been associated with decreased bleeding events,43,45,49 reduced need for additional medications,43,45,48,49 and improved HRQoL.50,51
Only one randomized clinical trial examining the efficacy of TPO-RAs in children has been conducted. In this phase 1/2 trial, pediatric patients with ITP for > 6 months were treated with romiplostim or placebo. Of the 17 patients who were randomized to receive romiplostim, 88% maintained a platelet count > 50 × 109/L for a median of 7 weeks compared with 0 patients in the placebo group (N = 5).52 Similar to adult patients, HRQoL was improved in subjects taking romiplostim.53
A subset of adult patients receiving TPO-RAs experienced sustained remission after the use of these agents.49,54 One rationale for this is that TPO-RAs have been shown to restore immune tolerance by improving Treg function,55 either as a direct effect of the drug or an indirect effect secondary to increased platelet number inducing immune tolerance by exposure to the platelet antigens.54,55
Primary safety concerns regarding TPO-RAs are related to the risk of BM reticulin formation and thrombosis. During the EXTEND study, annual BMs were performed on enrolled patients (N = 135) and none experienced grade 3 reticulin formation, symptoms related to BM dysfunction, or blast counts > 3% after 1 year of therapy.49 The causal relationship between TPO-RAs and BM reticulin remains unclear and the long-term consequences of increased reticulin formation are unknown. Prospective studies are ongoing to address these questions.
Thromboembolic events have been reported with both agents with long-term data, suggesting an event rate of 3.17-4.16 per 100 patient-years.48,49 It is difficult to ascertain the direct relationship between thrombosis and the use of TPO-RAs for several reasons. First, the majority of patients who experience an event have at least one additional risk factor for the development of thromboembolic events, such as hypertension, smoking, or diabetes.48,49 Secondly, the true baseline incidence of thrombosis associated with ITP and the role of antiphospholipid antibodies remains unknown. Lastly, thrombosis does not appear to be dependent upon drug-induced thrombocytosis and can occur at a low or normal platelet count.48,49
Unique to eltrombopag is the black box warning for hepatotoxicity. In the EXTEND study, 10% of patients met FDA criteria for drug-induced liver insufficiency, which required drug discontinuation in 6 patients. In the majority of patients (66%), the laboratory abnormalities resolved after discontinuation of the drug and, in some patients, the drug was restarted without encountering hepatotoxicity.49
Novel agents
Work is ongoing to develop novel therapies for patients with refractory ITP. In 2008, Podolanczuk et al presented pilot data on the oral Syk inhibitor R788 in adults with ITP.56 Syk is a downstream regulator of monocyte and macrophage phagocytosis57 and therefore the hypothesis was that blockage of the Syk pathway would effectively inhibit macrophage platelet destruction. In this pilot study (N = 16), half of the patients were able to demonstrate a sustained platelet count ≥ 50 × 109/L for the majority of visits. The most common side effect was GI symptoms including diarrhea, nausea, and emesis.
Knowledge that autoreactive CD4+ T cells are present in greater numbers in patients with ITP and that CD40 ligand (CD40L) is critical for T-cell–dependent B-cell expansion provided the rationale that anti-CD40L monoclonal antibodies could be therapeutic in ITP. The most recent investigation into anti-CD40L monoclonal antibodies (hu5c8 and IDEC-131) was undertaken in patients with chronic refractory ITP.58 Forty-six patients were treated hu5c8, with an overall response rate of 13%.34 After this, 31 patients were treated with IDEC-131 and had a similar overall response rate of 16%.34
Summary
Ongoing data are emerging that advance our comprehension of the pathogenesis of ITP and improve our understanding of the heterogeneity of the disease. With this growing body of literature, it is possible that we will be able to identify disease features and predictors of treatment response in the future that will help to guide management decisions. It is essential that clinical trials collect relevant ancillary biological samples to confirm existing data, understand the mechanism of action of medications, and identify predictors of response to various therapies. Ultimately, knowledge of disease biology coupled with information regarding response rates, patients' desires, medication side effects, and relevant patient-related outcomes will provide for individualized treatment plans.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests. Off-label drug use: rituximab and TPO agents.
Correspondence
Cindy Neunert, MD, MSCS, Georgia Regents University, 1120 15th St BG-2011, Augusta, GA 30912; Phone: 706-721-3626; Fax: 706-721-0264; e-mail: cneunert@gru.edu.