Abstract
After decades of virtually no progress, multiple myeloma survival has improved significantly in the past 10 years. Indeed, multiple myeloma has perhaps seen more remarkable progress in treatment and patient outcomes than any other cancer during the last decade. Recent data show that multiple myeloma is consistently preceded by a precursor state (monoclonal gammopathy of undetermined significance [MGUS]/smoldering multiple myeloma [SMM]). This observation provides a framework for prospective studies focusing on transformation from precursor disease to multiple myeloma and for the development of treatment strategies targeting “early myeloma.” This review discusses current biological insights in MGUS/SMM, provides an update on clinical management, and discusses how the integration of novel biological markers, molecular imaging, and clinical monitoring of MGUS/SMM could facilitate the development of early treatment strategies for high-risk SMM (early myeloma) patients in the future.
Introduction
Although monoclonal gammopathy of undetermined significance (MGUS) is commonly referred to as single entity in the literature, there are actually 2 kinds of MGUS: lymphoid (or lymphoplasmacytoid) MGUS and plasma cell MGUS.1 Approximately 15% to 20% of MGUS cases secrete IgM and most have a lymphoid or lymphoplasmacytoid phenotype. In contrast, most non-IgM (IgG > IgA > Ig light chain only > IgD > IgE) MGUS cases have a plasma cell phenotype. Typically, patients with plasma cell MGUS are at risk of progression to multiple myeloma or related plasma cell disorders, whereas patients with lymphoid MGUS may progress to Waldenström macroglobulinemia, lymphoma, or other malignant lymphoproliferative disorders.1,2 Furthermore, there is virtually no overlap of the molecular genetic events responsible for the molecular pathogenesis of the 2 kinds of MGUS, suggesting that they are quite distinct biological entities. Reflective of a higher burden of monoclonal plasma cells within the BM, smoldering multiple myeloma (SMM) is distinguished from MGUS by higher cutoff values while maintaining a lack of end-organ damage with conventional methods of assessment. This review focuses on plasma cell MGUS and SMM.
MGUS and smoldering myeloma: from 1978 to the 21st century
The 2 known precursors to multiple myeloma, MGUS and SMM, were first described by Kyle and Greipp in 1978 and 1980, respectively, as the presence of an M-protein in the serum and/or excess BM plasma cells in the absence of clinical evidence of either multiple myeloma or another lymphoproliferative disorder.3,4
In 2003, the International Myeloma Working Group (IMWG) developed the first consensus definitions of MGUS and SMM.5 MGUS was defined as the presence of serum M-protein < 3 g/dL with fewer than 10% monoclonal plasma cells in the BM; SMM was defined as either serum M-protein ≥ 3 g/L or ≥ 10% monoclonal plasma cells in the BM. In contrast to these laboratory-based definitions, a diagnosis of multiple myeloma is based on the clinical assessment of myeloma-related end-organ impairment in the presence of an M-protein and/or monoclonal plasma cells. In the 2003 IMWG criteria, end-organ damage was defined using both the classic “CRAB” criteria of hypercalcemia (serum calcium > 11.5 mg/dL), renal failure (defined by creatinine > 1.95 with no other etiology), anemia (hemoglobin < 10 g/dL), or skeletal lesions (lytic lesions by skeletal survey, osteoporosis with pathologic fractures, or cord compression) and additional criteria including recurrent bacterial infections (> 2 in 12 months), amyloidosis, or symptomatic hyperviscosity.5
In the updated 2010 IMWG diagnostic criteria, plasma cell MGUS was defined as serum M-protein < 3 g/dL, clonal plasma cell population in BM < 10%, and absence of end-organ damage (CRAB criteria of multiple myeloma; Table 1).6 The CRAB criteria were revised slightly in the 2010 version and include: hypercalcemia with calcium level > 11.5 mg/dL, renal insufficiency with serum creatinine > 2.0 mg/dL or estimated creatinine clearance <40 mL/min, normochromic normocytic anemia with a hemoglobin value < 10 g/dL (or a hemoglobin value < 2 g/dL below the lower limit of normal), and bone lesions (lytic lesions, severe osteopenia, or pathological fractures).6
Reflective of a higher burden of monoclonal plasma cells within the BM, SMM is distinguished from MGUS by higher cutoff values while maintaining a lack of end-organ damage. IMWG diagnostic criteria from 2010 established SMM as serum M-protein ≥ 3 g/dL and/or clonal plasma cell population in BM ≥ 10% and lack of end-organ damage (CRAB criteria).6 Based on retrospective data from the Mayo Clinic, risk of progression from SMM to multiple myeloma is 10% per year for the first 5 years, 3% per year for the next 5 years, and 1% for the subsequent 10 years; MGUS is associated with an average 1% annual risk of progression to multiple myeloma.7,8
From precursor disease to multiple myeloma: biological insights
Based on the prospective Prostate, Lung, Colorectal, and Ovarian (PLCO) Cancer Screening Trial, annual serum samples were collected from 77 469 healthy donors. Among 71 patients who during a 10-year follow-up time developed multiple myeloma, stored prediagnostic serum samples consistently demonstrated MGUS in the years before the malignant diagnosis.9
General understanding of tumor microenvironment interactions and genetic aberrations leading to multiple myeloma has prompted researchers to better characterize molecular and pathogenetic events surrounding multiple myeloma precursor disease. Based on current standard technologies (eg, FISH), the molecular makeup of myeloma precursor disease states and multiple myeloma are strikingly similar and no defining molecular features unique to multiple myeloma have been identified. In addition, the transformative genetic events that drive disease progression are unclear. For example, cytogenetic aberrations in multiple myeloma can be divided into 2 general entities with partially overlapping features: hyperdiploid (approximately 50%) and nonhyperdiploid (approximately 40%).10-14 The hyperdiploid group includes recurrent trisomies with 48-74 chromosomes.15 The nonhyperdiploid group (< 48 or > 74 chromosomes) is often associated with translocations involving the immunoglobulin heavy chain (IgH) locus at 14q32 and includes hypodiploid, near tetraploid, and pseudodiploid tumors.15 In a study using FISH, 72/189 (42%) MGUS, 70/127 (63%) SMM, and 223/338 (57%) multiple myeloma cases were found to be hyperdiploid.16 IgH rearrangements were found at similar prevalence rates among 78/189 (41%) MGUS, 44/125 (35%) SMM, and 183/398 (46%) multiple myeloma patients.16 Although the prevalence of chromosome 13 deletion is higher (50%) among multiple myeloma patients compared with MGUS patients (25%), studies suggest a higher frequency among patients with t(4,14) and t(4,16) rearrangements.13,16 In 2003, an international workshop assembled to review cytogenetic studies to evaluate whether MGUS and SMM cases have the same detectable anomalies that are often found in multiple myeloma.17 Indeed, p53 deletions and p18 deletions and mutations have been associated with aggressive, extramedullary multiple myeloma.15 Point mutations such as N-RAS, K-RAS, MYC up-regulation, and gain or loss of chromosome 1q or 1p also seem to correlate with disease progression from myeloma precursor disease of MGUS and SMM.18-22
Gene expression profiling (GEP) has been used to identify molecular signatures associated with different risks of progression from precursor disease to multiple myeloma. For example, in one study, 52 genes were investigated in plasma cells derived from healthy controls, MGUS, SMM, and multiple myeloma patients; the investigators showed that hierarchical clustering identified 4 groups from GEP analysis.21 However, these GEP groups have not yet been validated in an independent cohort of precursor patients and correlated to clinical outcome. GEP analyses of MGUS have inherent problems. The percentage of plasma cells is low (by definition < 10%), so that there is significant contamination with other kinds of cells despite selection of CD138+ cells on magnetic beads. In addition, in MGUS patients—unlike in multiple myeloma patients—monoclonal plasma cells are likely to be significantly contaminated with normal plasma cells (due to the relatively low percentage of monoclonal plasma cells in MGUS). Until we have better processing methods and better assays, one has to be cautious when interpreting GEP analyses of plasma cells selected by CD138 expression from MGUS patients.
In the classical view of transformation from MGUS/SMM to multiple myeloma, an initiating hit is required to immortalize a myeloma-propagating cell, which is then destined to acquire (through loss of heterozygosity, gene amplification, mutation, or epigenetic changes) additional genetic hits over time.23 Per this traditional view, these additional genetic hits further deregulate the myeloma-propagating cell and lead to the clinically recognized features of multiple myeloma.23 However, emerging insights based on the currently best available technologies suggest that there is substantial complexity in the genetic basis of multiple myeloma and its precursor states; standard technologies rather reflect the predominant clonal population and fail to take into account the presence of intratumoral subclonal heterogeneity.24,25 Using single nucleotide polymorphism–based mapping arrays, a progressive increase in the incidence of copy number abnormalities from MGUS to SMM and to multiple myeloma (median 5, 7.5, and 12 per case, respectively) has been observed recently (P = .006); gains on chromosome 1q, 3p, 6p, 9p, 11q, 19p, 19q, and 21q and 1p, 16q, and 22q deletions were significantly less frequent in MGUS than in multiple myeloma.26 Although multiple myeloma has more copy number abnormalities and copy number-neutral loss of heterozygosity than its precursor states, MGUS is as genetically aberrant as multiple myeloma and, based on current knowledge, the transition from MGUS to multiple myeloma does not appear to be associated with a particular chromosomal imbalance, but rather with an expansion of altered clones that are already present in MGUS.26 As proposed by Morgan et al,23 the heterogeneity observed in the transformation from MGUS/SMM to multiple myeloma is likely, from a Darwinian-selection perspective, to be the essential feature of clonal evolution, disease progression, and relapse (Figure 1). Based on this understanding, it is becoming increasingly plausible that, after disease initiation, the molecular events that are necessary for myeloma development are not attained in a linear fashion, but rather through branching, nonlinear pathways that are typical of those proposed by Darwin to explain the evolution of species.23 The idea of this model is that mutations are acquired randomly and are selected based on the clonal advantage they confer (Figure 1).27 Furthermore, as proposed by Morgan et al, if myeloma-propagating cell are the source of sustained myeloma growth, then they too should be genetically and epigenetically diverse to maintain a clonal advantage and to achieve the transitions between disease stages.23

Pathway to multiple myeloma. The transition of MGUS to plasma cell leukemia has been traditionally represented as a linear pathway (A). However, it is more likely that the pathway to myeloma is through branching pathways typical of those that are associated with the evolution of species (B). The key molecular events leading to disease evolution are represented as diamonds and indicate distinct patterns of driver mutations. This simple branching model clearly has implications for targeted treatment because the multiple distinct subclones could lead to differential responses to treatment. Reprinted with permission from Morgan et al.23
Pathway to multiple myeloma. The transition of MGUS to plasma cell leukemia has been traditionally represented as a linear pathway (A). However, it is more likely that the pathway to myeloma is through branching pathways typical of those that are associated with the evolution of species (B). The key molecular events leading to disease evolution are represented as diamonds and indicate distinct patterns of driver mutations. This simple branching model clearly has implications for targeted treatment because the multiple distinct subclones could lead to differential responses to treatment. Reprinted with permission from Morgan et al.23
Little is known about the epigenetic changes necessary for progression from MGUS/SMM to multiple myeloma.28 DNA can be modified by methylation of cytosine residues in CpG dinucleotides and chromatin structure may be modified by histone modifications such as methylation, acetylation, phosphorylation, and ubiquitylation.29 Both DNA and histone modifications can play a part in modulating gene expression.30 Based on recent data, the most important epigenetic changes during the transformation from MGUS to multiple myeloma are global DNA hypomethylation and gene-specific DNA hypermethylation.31 MicroRNA (miRNA) profiling has also yielded interesting results while further characterizing MGUS and SMM compared with multiple myeloma patients. miRNAs are noncoding, single-stranded RNA molecules known to influence various tumor behavior by regulating gene expression.32 Compared with healthy controls, MGUS and multiple myeloma patients seem to up-regulate miR-21, miR-106b, miR-181a, and miR-181b, all of which are involved in B- and T-cell lymphocyte differentiation and oncogene regulation.33 Based on our current knowledge, notable differences reveal up-regulation of miR-32 and the miR-17∼92 cluster among multiple myeloma patients not found in MGUS patients.33 Future studies are needed to further elucidate the role of miRNAs in multiple myeloma precursor disease and their precise role in progression to frank symptomatic multiple myeloma.
The BM microenvironment plays a key role in MGUS/SMM initiation and propagation.34 Although the BM microenvironment is commonly referred to as the “nontumor” entity, it has to be kept in mind that it is a complex network including a broad range of cells and factors. Indeed, the BM microenvironment consists of 3 components: the cellular component (hematopoietic and nonhematopoietic cells, including the vasculature); the extracellular matrix component (fibrous proteins, proteoglycans, glycosaminoglycans, and small integrin-binding ligand N-linked glycoproteins [SIBLING]); and the soluble component (cytokines, growth factors, adhesion molecules, and other factors).35 The 3 components are intertwined with multiple feedback loops within and between the compartments. Shared positive and negative interactions among a range of cells in the BM (such as: stromal cells, osteoclasts, osteoblasts, immune cells (T lymphocytes, dendritic cells), other hematopoietic cells and their precursors, and vascular endothelial cells34,35 ) are mediated by a variety of adhesion molecules, cytokines, and receptors. Additional stimuli such as hypoxia result in activation of HIF-1α and secretion of VEGF.36 In MGUS/SMM, there are multiple biological aspects that are affected by interactions between abnormal plasma cells and the BM microenvironment. Such aspects include homing to the BM, spread to secondary BM sites by the bloodstream, generation of paracrine factors (eg, IL-6, IGF-1, and APRIL), osteoclastogenesis, inhibition of osteogenesis, humoral and cellular immunodeficiency, angiogenesis, and anemia.34 Interestingly, mechanisms involved in, for example, homing, differentiation, and survival of abnormal plasma cells appear to be qualitatively similar to those of abnormal plasma cells; however, in MGUS/SMM, the composition of cells in the BM microenvironment is altered.34 It remains unknown whether the altered composition of cells in the BM microenvironment is due to abnormal plasma cells, if the altered composition of cells in the BM microenvironment precedes proliferation/activation of abnormal plasma cells, or if there is a combination of these mechanisms.
Clinical predictors of progression
Currently, the serum free light chain (sFLC) ratio is one of the most promising clinical biomarkers in asymptomatic myeloma. The sFLC ratio has been used as a prognostic indicator both in patients with MGUS37 and SMM.38 Presently, there is general consensus that BMPC levels ≥ 10%, serum M protein levels ≥ 3 g/dL, and abnormal sFLC ratios (≤ 0.125 or ≥ 8) increase the probability that SMM will develop into multiple myeloma.38,39 In one study, patients with non-IgG MGUS, an abnormal sFLC ratio (defined as < 0.26 or > 1.65), and a high serum M-protein level (> 1.5 g/dL) had a 58% absolute risk of disease progression after 20 years, whereas MGUS patients with ≤1 risk factors had only a 21% and 5% absolute risk of disease progression, respectively.37 In a separate study, SMM patients with a sFLC ratio ≥ 100 were found to develop multiple myeloma within a median of 15 months versus 55 months (median) for patients with a ratio < 100.39 In addition, normalization of sFLC ratios may lead to more favorable clinical outcomes independently of other variables.40 One study found that the sFLC ratio may be a good prognostic marker for determining which patients with high-risk SMM will receive the greatest benefit from early treatment.39 Given their findings, the investigators recommended initiating early treatment in patients with high-risk SMM and an sFLC ratio ≥ 100.41 Although plasma cells and sFLC have prognostic potential, they lack specificity and do not have enough positive predictive value to be useful in determining whether treatment should be initiated in patients with early myeloma.
Based on recent advances in immunophenotyping plasma cells and measuring sFLC, 2 independent risk stratification schemes for MGUS and smoldering myeloma have been designed by the Mayo Clinic8,37,38 and the Spanish PETHEMA Study Group.42 The Mayo Clinic criteria are primarily based on the levels of serum protein markers (serum protein electrophoresis [SPEP] with immunofixation and FLC assay). In a retrospective study of 1148 patients diagnosed with MGUS with long-term follow-up, M-protein > 1.5 g/dL, non-IgG MGUS, and an sFLC ratio < 0.26 or > 1.65 were independent risk factors for progression. At 20 years, patients with no risk factors had a 5% risk of progression compared with 21%, 37%, and 58% for patients with 1, 2, or 3 risk factors, respectively.37 Recently, a study screening for sFLC abnormalities without a detectable M-protein found a much lower risk of progression to multiple myeloma compared with conventional MGUS.43 In a study following 276 patients with SMM, M-protein ≥3 g/dL, BM plasma cells ≥10%, and an sFLC ratio deviating outside the nonstandard range of 0.125-8 were found to be independent risk factors for progression. At 5 years, risk of progression to multiple myeloma was 25%, 51%, and 76% for patients with 1, 2 or 3 risk factors, respectively.8,38 In contrast, the risk stratification scheme of the PETHEMA Study Group has focused on the use of multiparameter flow cytometry of the BM to quantify the ratio of abnormal, neoplastic plasma cells (aPCs) to normal plasma cells. At 5 years of follow-up, patients with MGUS with neither ≥ 95% aPCs nor DNA aneuploidy were found to have a very small, 2% risk of progression compared with a 10% risk for patients with one risk factor and a comparatively high 46% risk of progression at 5 years for patients with both.42 For patients with SMM, ≥ 95% aPCs and a reduction of uninvolved immunoglobulins were independent risk factors for progression, with rates of progression at 5 years being 4%, 46%, and 72% for patients with neither, one, or both risk factors, respectively.42 Recently, we conducted a prospective trial to assess the degree of concordance between these 2 models by comparing the distribution of SMM patients classified as low, medium, and high risk for progression.44 A total of 77 SMM patients were enrolled in our prospective natural history study. Per study protocol, each patient was assigned risk scores based on both the Mayo and the Spanish models. The Mayo Clinic model identified 38, 35, and 4 patients as low, medium, and high risk, respectively. The Spanish PETHEMA model classified 17, 22, and 38 patients as low, medium, and high risk, respectively. There was significant discordance in overall patient risk classification (28.6% concordance) and in classifying patients as low versus high (P < .0001), low versus non-low (P = .0007), and high versus non-high (P < .0001) risk.44 Although this study currently has limited follow-up data, the observed discordance between the 2 clinical models suggests that the identification and validation of other biomarkers will be needed before clinicians can determine whether initiation of early treatment is beneficial to patients with high-risk SMM.
Finally, suppression of uninvolved immunoglobulins in MGUS as detected by suppression of the isotype-specific heavy and light chain (HLC-pair suppression) was recently assessed in relation to risk of progression to multiple myeloma. Using baseline serum samples (available for 999 persons) obtained within 30 days of an MGUS diagnosis at Mayo Clinic (1960-1994), quantitation of individual heavy/light chains (for example, IgGλ in IgGκ MGUS patients) was conducted.45 This study identified HLC-pair suppression in 27% of MGUS patient samples compared with 11% of patients with suppression of uninvolved IgG, IgA, or IgM. HLC-pair suppression was a significant risk factor for progression (hazard ratio [HR] = 2.3; 95% confidence interval [CI], 1.5-3.7; P < .001). On multivariate analysis, HLC-pair suppression was an independent risk factor for progression to malignancy in combination with serum M-spike concentration, heavy chain isotype, and sFLC ratio (HR = 1.8; 95% CI, 1.1-3.00; P = .018). The finding that HLC-pair suppression predicts progression in MGUS and occurs several years before malignant transformation has implications for myeloma biology.45
Despite the above-mentioned limitations of currently available risk-factor models, the most recently updated 2010 IMWG guidelines6 propose the following clinical management of individuals diagnosed with MGUS and SMM.
Current clinical management strategies
In 2013/2014, the cornerstone of managing MGUS/SMM involves a prudent “watch and wait” strategy. Outside of clinical trials, there are no current standardized treatment options for MGUS or SMM. Aggressive disease monitoring is based on whether patients fit into MGUS or SMM precursor disease and the above outlined risk factors in the Mayo Clinic model and Spanish study group model.6 For the first time, the 2010 IMWG guidelines suggest risk stratifying all patients with MGUS and SMM and differentially monitoring patients on the basis of their risk category.6 Importantly, the recommendations state that patients with low-risk MGUS (∼ 50% of all MGUS cases) by the Mayo Clinic criteria (IgG M-protein < 1.5 g/dL with a normal sFLC ratio) in the absence of concerning symptoms such as anemia or poor renal function, no further initial evaluation is needed. Subsequently, low-risk MGUS patients should be followed with SPEP, CBC, calcium, and creatinine at 6 months and, if stable, every 2 to 3 years after that.6 As an alternative strategy, the 2010 IMWG suggests that check-up of low-risk MGUS only be performed when symptoms for multiple myeloma arise, thus abrogating the need for scheduled long-term follow-up in stable patients.6,46
In contrast to low-risk MGUS, the 2010 IMWG guidelines state that MGUS patients with any risk factor (ie, intermediate- or high-risk MGUS) should be evaluated with baseline BM examination with cytogenetics and FISH studies in addition to bone imaging studies such as skeletal surveys.6 Intermediate- and high-risk MGUS patients should be followed with an SPEP every 6 months for the first year, followed by annual SPEP and routine laboratory tests.6
Given their increased risk of progression, the 2010 IMWG guidelines state that in SMM patients, an SPEP and physician visit should be repeated every 2 to 3 months for first year, followed by every 4 to 6 months for 1 year, with eventual 6- to 12-month evaluations if clinically stable thereafter.6 In SMM, beyond mandatory baseline BM examination and skeletal survey, the guidelines recommend magnetic resonance imaging (MRI) of the spine and pelvis because it can detect occult lesions, which, if present, predict for a more rapid progression to multiple myeloma.6,47
It is critical to recognize that in a disease such as multiple myeloma, in which defining criteria rely on the presence or absence of end-organ damage, diagnosis is only as good as the tools and technology able to detect end-organ damage. Although researchers strive to improve upon the available diagnostic armamentarium, clinical acumen on the part of the physician should be emphasized while playing a central role in disease monitoring. For example, in SMM or high-risk MGUS patients highly suspicious as harboring bone disease, imaging evaluation may be better served by obtaining MRI or positron emission tomography (PET)–computed tomography (CT) rather than traditional skeletal surveys. MGUS or SMM patients with unexplained anemia or renal disease should be evaluated for other underlying causes and with complete BM examination including cytogenetics and FISH studies.
As already discussed in part (see “Clinical predictors of progression”), recent studies suggest that additional features such as BM plasmacytosis ≥ 60%48 ; an abnormal FLC-ratio ≥ 100 (involved kappa) or < 0.01 (involved lambda)39 ; and/or focal BM lesions detected by functional imaging including PET-CT and/or MRI47,49 in asymptomatic individuals may warrant a clinical diagnosis of multiple myeloma. Based on expert discussions at the IMWG meeting in Stockholm in June 2013, it is anticipated that updated consensus criteria will be defined in the near future (Table 1).
From X-ray to molecular imaging
The definition of multiple myeloma precursor disease is based on the lack of end-organ damage, including bone lytic lesions. According to the most recent consensus guidelines of the IMWG updated in 2011, radiological skeletal survey is still the gold standard for the initial workup of patients with multiple myeloma.50 This means that most of the data from clinical trials are based on the definition of the presence of bone disease as detected by conventional X-ray. Skeletal survey has several advantages. For example, it is widely available and comparatively cheap. With current digital scanners, the radiation dose is low (about 3-4 mSv). However, conventional X-ray has also some important drawbacks. First of all, it has been known for a long time that 30% to 50% of the bone mass has to be destroyed before conventional radiography is able to detect the damage.51 This means that if myeloma is seen as continuous course of disease from MGUS to SMM to symptomatic disease, the occurrence of bone destruction is a relatively late event.52 Multiple myeloma patients who often present with severe bone pain have to move their limbs to place them on the films and the whole examination therefore takes a relatively long time. Some parts of the skeleton project one upon the other if the patient is placed in the linear projection of the path of rays. It is not infrequent that intestinal gas, especially if located in front of the osseous pelvis, mimics osteolyses.
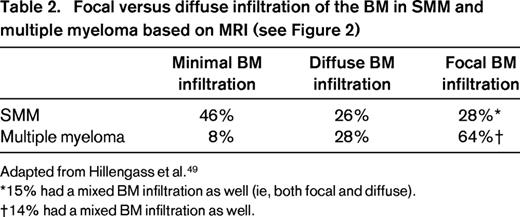
New imaging techniques allow detection of myeloma manifestations earlier than conventional radiography.49 Although CT and skeletal survey detect lytic lesions in the bone as a secondary event, MRI is able to assess the disease in the BM itself independently from the growth pattern and therefore can provide information on the actual tumor burden. In addition to those functional techniques such as PET/CT or PET/MRI, dynamic contrast-enhanced MRI and diffusion weighted imaging MRI comprise the possibility of gaining information regarding disease activity.49 Interestingly, Hillengass et al reported recently that 30% of patients with SMM have BM infiltration patterns similar to multiple myeloma when using whole-body MRI (Figure 2, Table 2).49,53 Indeed, based on 149 SMM patients followed up to 60 months, the investigators found SMM patients with > 1 focal BM lesion to have a significantly (P < .001) shorter time (median time to progression: 13 months vs not reached) to develop multiple myeloma compared with SMM patients with ≤1 focal BM lesion.47

Development of early treatment strategies
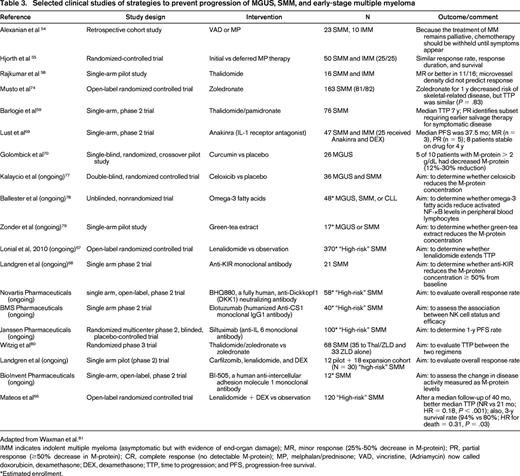
Among the first studies examining treatment of SMM was a 1988 retrospective study of 23 patients with SMM and 10 patients with lytic lesions but no symptoms. These patients were treated with 1 of 2 chemotherapy regimens, but the study failed to show a statistically significant difference in the end points of remission or survival.54 Because this study was limited in its size and design, a randomized controlled trial of initial (at diagnosis) versus delayed (at symptoms) treatment with melphalan-prednisone for 50 patients with SMM or indolent multiple myeloma (asymptomatic disease but with evidence of end-organ damage) was performed, finding no difference in response rate, response duration, or survival (Table 3).55 Based on these observations, for several years, it has been deemed inappropriate to recommend that patients undergo a difficult chemotherapy regimen with no evidence of a clinical benefit. However, both of these studies were performed when the best available treatment for multiple myeloma was melphalan-prednisone, a regimen with a poor therapeutic index for treatment of an asymptomatic condition.
The development of modern therapies with improved efficacy and less toxicity have renewed interest in the treatment of SMM. Thalidomide is a noncytotoxic drug used to treat refractory multiple myeloma that has been shown to have antiinflammatory, antiangiogenic, and immunomodulatory properties.56,57 In 2001, a trial of thalidomide as treatment for SMM in 16 patients was performed (Table 3). Partial response was achieved in 38% of patients.58 These data may be confounded by the inclusion of patients with indolent multiple myeloma, who would now be classified as having multiple myeloma. In 2008, a single-arm phase 2 trial including 76 patients with SMM treated with thalidomide and pamidronate did not show a clear overall benefit to treatment (Table 3). Paradoxically, patients who initially displayed at least a partial response to thalidomide had a shorter median time to treatment (< 2 years) than patients who showed no improvement (not reached in 8 years).59 Although it remains to be proven, the investigators speculate that this may be due to a greater initial response in patients with more proliferative tumors or selection of aggressive clones due to treatment. This study was further complicated by poor tolerance to thalidomide due to peripheral neuropathy and dizziness resulting in discontinuation in greater than half of patients.59 These dose-limiting toxicities have prompted the use of less toxic drugs that share mechanistic features with thalidomide.57 Lenalidomide has proven efficacy with dexamethasone in both relapsed-refractory and newly diagnosed multiple myeloma.57,60-64 Although the side effect profile was improved relative to thalidomide, BM suppression and venous thromboembolism remained as significant adverse events.61,65 The first phase 3 clinical trial using novel drugs (lenalidomide-dexamethasone vs clinical surveillance) in SMM was published in 2013 (Table 3).66 After a median follow-up of 40 months, the median time to progression was significantly longer in the treatment group than in the observation group (median not reached vs 21 months; HR for progression = 0.18; 95% CI, 0.09-0.32; P < .001). The 3-year survival rate was also higher in the treatment group (94% vs 80%; HR for death = 0.31; 95% CI, 0.10-0.91; P = .03). A partial response or better was achieved in 79% of patients in the treatment group after the induction phase and in 90% during the maintenance phase. Toxic effects were mainly grade 2 or lower. These data serve as proof of principle that the treatment of high-risk SMM can be accomplished without excessive toxicity and may delay progression to multiple myeloma. We do not know if there will be differences between the 2 study arms with regard to quality of life. Finally, we do not know whether patients in the lenalidomide-dexamethasone treatment arm who later will develop multiple myeloma may have an altered susceptibility to therapy. These are very important questions that need to be addressed as soon as follow-up data are mature and allow formal, sufficiently powered statistical analysis.
During the past few years, new SMM treatment studies have opened in the United States. For example, the Eastern Cooperative Oncology Group (ECOG) and Southwest Oncology Group (SWOG) study groups in North America have initiated a collaborative, randomized phase 3 study designed to compare lenalidomide alone (versus clinical surveillance) in SMM patients.67 Recently, several monoclonal antibody–based studies have been developed for SMM patients. For example, anti-DDK1, anti-CS-1, anti-IL6, anti-intercellular adhesion molecule 1, and anti-KIR (Table 3)68 monoclonal antibodies have been and are being used (Table 1). Results from these studies are currently pending. Very recently, a pilot study using lenalidomide-dexamethasone in combination with carfilzomib was opened at the National Cancer Institute, National Institutes of Health (NCI/NIH) in Bethesda, and the first preliminary results from this study were presented at the IMWG meeting in 2013 in Kyoto, Japan (Table 3). Based on small numbers, the best overall response rate was 100% and the best near complete remission/complete remission rate was 75% (Table 1). The results from several of these ongoing trials are expected to become available in the near future.
Finally, a few smaller trials have studied other interventions with benign side effect profiles aimed at limiting the progression of SMM or MGUS, including anakinra, a targeted IL-1 receptor antagonist69 ; curcumin, a traditional Indian spice with preclinical antimyeloma activity70-73 ; and bisphosphonates, believed to block the initial formation of lytic lesions and alter the BM microenvironment (Table 3).74,75 For example, in 2008, a randomized study compared zoledronic acid versus surveillance in SMM and demonstrated reduced skeletal events in the treatment arm (zoledronic 55.5% vs surveillance 78.3%; P = .041); however, there was no difference in median time to progression (P = .83) to full-blown multiple myeloma.74 At this time, none of these studies has had sufficient power or study design to significantly alter clinical practice. Some data are further limited by use of nonstandard end points and response criteria.76 Despite these limitations, these trials are crucial in their ability to open avenues for further research. Selected published and ongoing clinical trials studying treatment of MGUS/SMM are listed and described in Table 1.
Summary and future directions
As stated above, based on the IMWG 2010 guidelines, patients diagnosed with MGUS and SMM should not be treated outside of clinical trials.6 In standard clinical practice, SMM patients shall receive close follow-up with repeat laboratory testing at 2 to 3 months during the first year and, if stable, every 4 to 6 months thereafter due to the high initial risk of progression.6
Overall, treatment trials for MGUS patients are complicated because these individuals are relatively healthy and the majority have a low lifetime risk of progression, especially when other causes of death are taken into account.7 Therefore, it seems reasonable to propose that an ideal treatment would be very effective, nontoxic, and directed toward patients with high risk of progression. We are nearing this end point, but fundamental unanswered questions remain.
The high rate of progression of SMM into symptomatic disease makes the idea of an early treatment strategy attractive to patients and clinicians. Several promising clinical trials are already in progress both in Europe and in the United States (Table 1). In the future, novel agents currently used to treat relapsed or refractory multiple myeloma may be applied in clinical trials designed for SMM patients. Newer drugs with more favorable side effect profiles may have the potential to be used in the treatment of SMM without the often treatment-terminating adverse effects of thalidomide. The time may be right for the commencement of clinical trials of these novel agents in the treatment of SMM, but such studies should be careful to analyze key end points such as time to progression and overall survival because past experience has indicated that partial and complete responses may not be illustrative of benefit. As research moves forward in characterizing precursor disease, it is important to tread carefully in clinical trials involving treatment of an asymptomatic disease state.
In summary, although current evidence does not support the treatment of SMM outside of clinical studies, it seems reasonable to support the development of early treatment trials that integrate molecular monitoring. Future goals should be: (1) to provide molecularly based predictions of transformation from MGUS/SMM to multiple myeloma, (2) to use molecular markers to counsel patients with regard to future clinical follow-up/intervention and to assess health-related quality-of-life factors to capture the overall effect of therapy, and (3) to develop novel treatments for patients with high-risk SMM (“early myeloma”) with the aim of delaying progression and potentially offering cure.
Acknowledgments
This work was supported by the Intramural Research Program of the National Cancer Institute of the National Institutes of Health.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests. Off-label drug use: Treatment of SMM: ongoing clinical trials (lenalidomide, carfilzomib).
Correspondence
Dr Ola Landgren, Multiple Myeloma Section, Metabolism Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, 9000 Rockville Pike, Bldg 10/Room 13N240, Bethesda, MD 20892; Phone: 301-496-0670; Fax: 301-496-9956; e-mail: landgreo@mail.nih.gov.