Abstract
The discovery of the JAK2 V617F mutation in the classic BCR-ABL1–negative myeloproliferative neoplasms in 2005 catalyzed a burst of research efforts that have culminated in substantial dividends for patients. Beyond JAK2 V617F, a more detailed picture of the pathobiologic basis for activated JAK-STAT signaling has emerged. In some patients with myelofibrosis (MF), next-generation sequencing technologies have revealed a complex clonal architecture affecting both genetic and epigenetic regulators of cell growth and differentiation. Although these bench-top findings have informed the clinical development of JAK inhibitors in MF, they have also provided scientific context for some of their limitations. The JAK1/JAK2 inhibitor ruxolitinib is approved for treatment of MF in North America and Europe and other lead JAK inhibitors discussed herein (fedratinib [SAR302503], momelotinib [CYT387], and pacritinib [SB1518]), have entered advanced phases of trial investigation. Uniformly, these agents share the ability to reduce spleen size and symptom burden. A major challenge for practitioners is how to optimize dosing of these agents to secure clinically relevant and durable benefits while minimizing myelosuppression. Suboptimal responses have spurred a “return to the bench” to characterize the basis for disease persistence and to inform new avenues of drug therapy.
Lessons from the bench
Normal hematopoiesis relies on the cytoplasmic tyrosine kinase JAK2, which associates with the type I cytokine receptors for erythropoietin, thrombopoietin, and G-CSF.1 In 2005, several groups identified JAK2 V617F (V617F) as a highly recurrent somatic mutation in patients with polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF).2-5 Aggregate studies indicate a V617F frequency of 95% to 98% in PV and 50% to 60% in ET and PMF.6 V617F is located within the pseudokinase (JH2) domain of JAK2. Initial models predicted that the mutation relieves inhibition of the kinase (JH1) domain by the pseudokinase domain.7 However, recent crystal structure data indicate that the pseudokinase domain can act as a kinase that binds ATP and that key conformational interactions between JH2 residues F595 and V617F facilitate activation of the JH1 domain.8-10 The current ATP-competitive JAK inhibitors do not discriminate between mutant and wild-type JAK2, which may affect their therapeutic index. Such modeling information is critical to the development of next-generation mutant-specific inhibitors, which may exhibit better targeting of the malignant clone with less suppression of normal hematopoiesis.
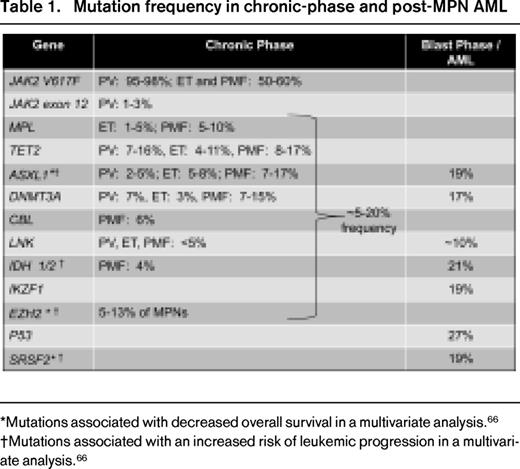
Dysregulated JAK-STAT signaling leads to increased proliferation and/or decreased apoptosis of neoplastic cells without the requirement for cytokine receptor binding by ligand. Although activation of the JAK-STAT pathway is common in myeloproliferative neoplasms (MPNs), it cannot be solely explained by the V617F or exon 12 JAK2 mutations that are found in ∼ 1% to 3% of PV patients.11,12 Other molecular alterations that interdigitate with this axis also contribute to MPN pathogenesis. For example, transmembrane domain mutations (eg, W515 L/K) in MPL,13,14 the receptor for thrombopoietin, are found in 1% to 5% of patients with ET and in 5% to 10% of PMF patients and lead to cytokine-independent growth and constitutive JAK-STAT signaling. Impaired negative regulation of JAK-STAT signaling due to mutations in LNK or CBL (< 5% frequency)15,16 or abrogation of suppressor of cytokine signaling (SOCS) activity (due to SOCS gene hypermethylation or increased protein phosphorylation) provide alternative explanations for perturbation of this pathway or for potentiation of mutant JAK2 activity itself.17,18
Although V617F is sufficient to produce a myeloproliferative phenotype in murine models,19-21 the acquisition of multiple pre- and post-V617F mutational events associated with different biologic pathways is common in MPN (Table 1)6,22 and contributes to disease initiation and progression. Data to support that V617F may arise on a preexisting abnormal clonal substrate derive from the observation that, in some patients, the V617F burden is relatively small compared with the proportion of cells with a coexistent karyotypic abnormality and that V617F can no longer be detected in a moderate proportion of acute myelogenous leukemia (AML) cases arising from a V617F-positive MPN.23 In chronic myeloid leukemia (CML), BCR-ABL1 is sufficient as a genetic driver and has served as a paradigmatic target of successful therapy with ABL tyrosine kinase inhibitors. In contrast, the finding that V617F is often accompanied by additional genetic changes, especially in advanced MPN/myelofibrosis (MF), has moderated expectations of deep clinical remissions with JAK inhibitor monotherapy. Abnormalities in epigenetic pathways that control DNA methylation and modification of chromatin (detailed by Abdel-Wahab24 ) and mutations in spliceosome factors have also emerged as important aspects of MPN pathogenesis, creating both challenges and opportunities for drug development.
Among the MPNs, MF is notoriously characterized by a pathologic milieu of proinflammatory and profibrotic cytokines (eg, IL-6, basic FGF, TGF-β, TNF-α, hepatocyte growth factor, VEGF, and others).25,26 These cytokines are implicated in MF-related constitutional symptoms and disease-related cachexia. Increased cytokine blood levels have been correlated with transfusion dependence, increased splenomegaly, thrombocytopenia, and shortened survival.26 In addition, their production is linked to various signaling networks, including JAK1 and JAK2.25,26 Therefore, the use of small molecules with anti-JAK1 and anti-JAK2 activity that inhibit inflammatory signaling cascades and V617F-bearing neoplastic cells is a rational application of these drugs. In this regard, cytokine down-modulation by ruxolitinib25 and fedratinib27 has been demonstrated. It has also been shown that cytokines secreted by BM stromal cells protect MPN clones from JAK2 inhibitor treatment,28 highlighting the need to target both the BM microenvironment and malignant clone.
Lessons from the bedside
In MF, the decision regarding when and what type of therapy to initiate depends on a nexus of factors related to the patient, disease stage,29 primary clinical manifestations, drug-specific considerations, and treatment goals (Figure 1). Similarly, after treatment is initiated, a nuanced understanding of the relationship among drug dose, hematologic and nonhematologic adverse events, and efficacy outcomes is critical to optimizing patient care. The experience with JAK inhibitors highlights many of these interacting issues.

Reduction of splenomegaly and symptoms are core benefits of JAK inhibitors
Splenomegaly.
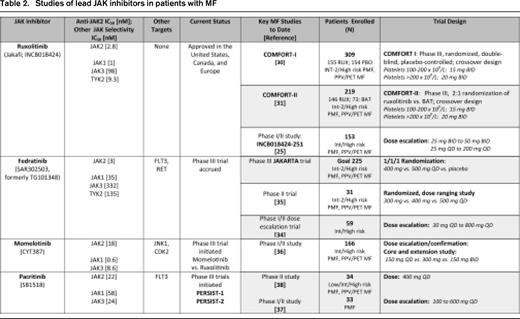
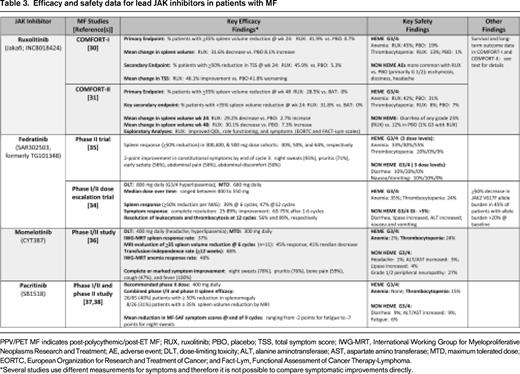
The extramedullary hematopoiesis associated with splenomegaly is a cardinal feature of MF and is linked to abdominal pain and impairment of quality of life (QOL). A ≥ 35% reduction in spleen volume by CT/MRI has been validated as a radiographic surrogate of an ∼ 50% reduction in palpable spleen length and has therefore been adopted in trials to quantify spleen changes. In the COMFORT-I trial (Tables 2, 3), 41.9% of patients in the ruxolitinib arm compared with 0.7% in the placebo group (P < .001) achieved the primary end point of a ≥ 35% reduction in spleen volume at week 24.30 The mean reduction in spleen volume was 31.6% with ruxolitinib compared with a mean spleen volume increase of 8.1% with placebo. In COMFORT-II, at week 48 (primary end point) and week 24 (key secondary end point), 28.5% and 31.8% of ruxolitinib-treated patients, respectively, achieved a ≥ 35% reduction of spleen volume versus 0% in the best available therapy (BAT) arm (Tables 2, 3).31 In both trials, almost all ruxolitinib patients experienced some reduction of spleen size, with a mean spleen volume reduction of ∼ 30%. Among the ruxolitinib-treated patients who achieved at least a 35% reduction in spleen volume in the COMFORT studies, ∼ 60% of patients maintained this response with follow-up ranging from 84 (COMFORT-II)32 to 112 weeks (COMFORT-I).33 A post hoc analysis of the COMFORT-I trial revealed that crossover patients demonstrated a smaller net reduction in spleen volume than patients originally randomized to ruxolitinib because of the interval spleen growth while they were receiving placebo before starting ruxolitinib.33
Similar reduction of spleen size (by either palpation or volumetric imaging) has been observed in phase 1/2 trials of fedratinib, momelotinib, and pacritinib (Tables 2, 3).34-38 With the caveat of differences in trial design/eligibility, results from the phase 3 trials of these drugs will help better define their comparative effects on the magnitude and durability of spleen reduction.
Symptoms.
Conventional agents such as hydroxyurea, immunomodulatory drugs (eg, thalidomide, lenalidomide, and pomalidomide) with or without corticosteroids, androgens, and erythropoiesis-stimulating agents have shown minimal effects in alleviating MF-related fatigue or other constitutional symptoms. In addition, until the development of the MF symptom assessment form,39 no instrument of patient reported outcomes (PRO) effectively canvassed the unique spectrum of symptoms afflicting MF patients. In phase 1/2 studies of ruxolitinib, fedratinib, momelotinib, and pacritinib, complete or marked improvement of the common MF constitutional symptoms was achieved in the majority of subjects.25,34-38 In the phase 3 COMFORT trials, the total symptom score (TSS), derived from a modified version of the MF symptom assessment form, was used to sum patient-reported individual scores for night sweats, itching, abdominal discomfort, pain under the ribs on the left side, early satiety, and muscle or bone pain.30,31 In COMFORT-I, a significantly higher proportion of patients in the ruxolitinib arm compared with the placebo group reported a ≥50% reduction in the TSS from baseline to week 24 (45.9% vs 5.3%; P < .001).30 Patients receiving ruxolitinib had a mean improvement of 46.1% in the TSS at week 24, whereas patients receiving placebo had a mean worsening of 41.8% (P < .001). Improvement in each individual symptom was observed, whereas all symptoms worsened in the placebo group.30 These findings mirrored significant improvements in other validated PROs that were also incorporated into the phase 3 trials.30,31,40 The degree of spleen volume reduction with ruxolitinib was correlated with improvements in the TSS and other PROs.40 For example, ruxolitinib-treated patients who achieved a ≥ 35% reduction in spleen volume experienced the greatest improvements in these PROs. Improvements in symptoms and QOL were sustained with 96 weeks of follow-up.33 Compared with placebo, ruxolitinib improved the TSS regardless of the degree of baseline splenomegaly, symptom burden, or other disease characteristics.30,41
Impact of JAK2 V617F mutation status on response
Given the successful application of ABL-tyrosine kinase inhibitors in CML, it is not surprising that some maintain the perception that JAK2 inhibitors, as an iteration of “targeted therapy,” only benefit patients with the JAK2 V617F mutation. However, JAK inhibitors do exhibit activity in V617F-negative patients, albeit the mechanism(s) are not well understood.
Rates of response or magnitude of spleen and symptom changes between V617F-positive and V617F-negative MF patients were not statistically different in the COMFORT trials,30,31 but favored V617F-positive patients. In COMFORT-I, the mean changes in spleen volume among ruxolitinib-treated V617F-positive and V617F-negative patients were −34.6% and −23.8, respectively.30 Similar trends were observed in the percentage reduction in TSS: −52.6% (V617F-positive) compared with −28.1% (V617F-negative).30 In the COMFORT-II trial, the rate of spleen volume reduction ≥ 35% was 33% versus 14% in ruxolitinib-treated V617F-positive and V617F-negative subgroups, respectively.31
Dose-response relationships
In COMFORT-I and COMFORT-II, a minimum platelet count of 100 × 109/L was required for eligibility, and starting doses of ruxolitinib depended on the platelet count at baseline (100-200 × 109/L: 15 mg twice daily [BID]; > 200 × 109/L: 20 mg BID). In COMFORT-I, ∼ 70% of patients underwent predefined dose adjustments during the first 12 weeks of therapy (primarily for cytopenias).30 By week 24, the median final titrated dose was 10 mg BID for the cohort starting at 15 mg BID, and remained 20 mg BID for the 20 mg BID starting cohort. Doses of 10 mg BID and higher were associated with a more robust level of treatment benefit.30,42 For example, TSS improvements of 60% to 71% were observed by week 24 with doses of 10 to 25 mg BID compared with only a 18% improvement in patients with a final titrated dose of 5 mg BID.42 A similar efficacy distinction between 5 mg BID and doses of 10 mg BID or higher was observed for spleen volume reduction: at 5 mg BID, the median spleen volume reduction of ∼ 10% was comparatively less than 31% to 41% median reductions in spleen volume in doses ranging from 10 to 25 mg BID.42 Given the proportion of patients undergoing dose reduction and/or interruption (77% of patients with baseline platelet counts of 100 to 200 × 109/L and 39% of those with baseline platelet counts > 200 × 109/L),41 there may be utility in alternative dosing strategies with ruxolitinib that are applicable to other JAK inhibitors. For example, a “start low and escalate” rather than “start high and de-escalate” algorithm may be prudent in some patients with lower blood counts.
Managing anemia and thrombocytopenia
Anemia and thrombocytopenia are expected “on-target” effects of JAK2 inhibition because of the dependence of both erythropoietin and thrombopoietin receptors on signaling via the JAK2 tyrosine kinase. In the COMFORT-I and COMFORT-II studies, anemia and thrombocytopenia (including grade 3/4 events) were more common with ruxolitinib (Table 1) than with placebo or BAT and typically peaked during the initial 8 to 12 weeks of therapy.30,31 During this time window, mean hemoglobin levels declined by ∼ 1 to 1.5 g/dL and, by week 24, increased to a higher steady state that was closer to the baseline hemoglobin. Higher-grade anemia or thrombocytopenia rarely led to discontinuation of ruxolitinib and was managed with brief treatment interruptions and dose modifications or packed RBC transfusions. Patients with new grade 3/4 anemia experienced symptomatic improvement and reductions in spleen volume similar to patients without anemia. With longer-term follow-up, the incidence of grade 3 or 4 anemia and thrombocytopenia decreased to levels observed with placebo treatment before crossover.30 Similarly, by week 36, the proportion of ruxolitinib-treated patients receiving RBC transfusions decreased to levels observed with the placebo group and thereafter remained stable.
Emergent grade 3-4 thrombocytopenia has been observed with other JAK inhibitors in phase 1/2 trials (both fedratinib and momelotinib: 24% frequency; pacritinib 15%; Tables 2, 3), is generally dose dependent, and may be influenced by baseline platelet count.34-38 In studies of fedratinib and pacritinib, spleen and symptom responses in patients with starting platelet counts in the range of 50 to 100 × 109/L have been commensurate with responses in patients with initial platelet counts > 100 × 109/L.37,38,43 In a ruxolitinib study enrolling subjects with low baseline platelet counts (50-100 × 109/L), most patients initiated at a dose of 5 mg BID were able to achieve a final dose at 24 weeks of 10 mg BID or higher.44 Approximately one-third of patients experienced a > 50% improvement in TSS and/or a ≥ 35% reduction in spleen volume.
Grade 3-4 anemia (35%) was observed in the phase 1/2 trial of fedratinib, with a similar incidence (30%-55%) observed in a follow-up phase 2 randomized study of doses ranging from 300 to 500 mg daily.35 In the three-arm phase 3 trial of this agent, doses of 400 versus 500 mg are being evaluated against placebo to determine the dose that maximizes benefit while minimizing myelosuppression. Higher-grade anemia with pacritinib appears to be less common.37,38
Momelotinib has garnered interest because of its erythroid-remitting activity. In the latest update of the phase 1/2 momelotinib study of 166 patients,36 the transfusion independence rate (minimum of 12 weeks) was 68% among 68 evaluable patients who exhibited baseline transfusion dependence. The median duration of RBC transfusion independence had not yet been reached. Among the dose cohorts of 150 mg once daily, 300 mg once daily, and 150 mg BID, the highest rate of transfusion independence was observed in the 300 mg once daily cohort.36
In addition to discretionary use of dose interruptions and modifications and packed RBC transfusions, combination strategies with erythropoiesis-stimulating agents, androgens such as danazol, and immunomodulatory agents are being evaluated for their ability to mitigate JAK-inhibitor–induced anemia.
Potential disease/survival modification with ruxolitinib
The COMFORT-I study was designed as an intention-to-treat analysis that included a crossover design with overall survival (OS) as a secondary end point.30 At the time of primary data cutoff, 10 deaths occurred in the ruxolitinib arm (6.5%) compared with 14 deaths in the placebo group (9.1%; hazard ratio [HR] = 0.67, P = .33). Although this difference was not statistically different, a planned analysis with 4 months of additional follow-up (median follow-up, 51 weeks) revealed a statistical difference in median OS: deaths were recorded in 13 patients (8.4%) in the ruxolitinib arm and 24 patients (15.7%) in the placebo arm (HR = 0.50, P = .04). This OS advantage persists with 2 years of median follow-up (27 vs 41 deaths in the ruxolitinib vs placebo groups, respectively; HR = 0.58, P = .03).33 In the COMFORT-II trial,31 which also followed an intent-to-treat design, no difference was found in the end point of OS at week 48. However, the COMFORT-II trial was amended to permit follow-up of patients during an extension phase of treatment. During this extension phase, 73% of patients originally assigned to the ruxolitinib arm and 62% patients randomized to the BAT arm were receiving ruxolitinib. After a median follow-up of 112 weeks, 20/146 (14%) and 16/73 (22%) deaths occurred in the ruxolitinib and BAT arms, respectively, suggesting an OS advantage favoring ruxolitinib (HR = 0.51, P = .041).32 Recently reported 3-year follow-up data from COMFORT-II indicate maintenance of the survival advantage with ruxolitinib with a 52% reduction in risk of death in the ruxolitinib arm compared with BAT (P = .009). The estimated probability of being alive at 144 weeks was 81% in the ruxolitinib arm and 61% in the BAT arm.45
In the absence of hematologic remissions, significant reduction in BM fibrosis and/or V617F allele burden, nor any proven modification of leukemia-free survival, other factors may explain the emerging survival advantage with ruxolitinib. Foremost is the enhancement of performance status related to reduction of splenomegaly and improvement of constitutional symptoms. In the phase 1/2 study of ruxolitinib,25 patients who experienced a ≥ 50% reduction in palpable spleen length from baseline had significantly prolonged survival compared with patients with a < 25% reduction in spleen size. Accordingly, patients who demonstrated an intermediate reduction in splenomegaly exhibited an intermediate survival. Post hoc analysis of the COMFORT-I trial showed a statistically prolonged survival in patients whose maximum weight gain or increase in total cholesterol were above the median for these variables compared with patients with lesser degrees of improvement.46 Improvement of constitutional symptoms, the presence of which is an adverse factor in current prognostic scoring systems, may also contribute to improved outcomes. These data suggest that the survival advantage associated with ruxolitinib may be partly explained by reversion of the catabolic state associated with MF. Data regarding survival outcomes are not yet available with other agents.
Dose duration and intensity: potential impact on long-term outcomes
Different long-term outcomes have been reported by the Mayo Clinic and the MD Anderson Cancer Center (MDACC) in follow-up of their own institutional cohorts enrolled in the phase 1/2 study (INCB18424-251) of ruxolitinib. Including adjustment for the Dynamic International Prognostic Scoring System score, the Mayo Clinic reported no significant difference in the survival rate of their 51 ruxolitinib-treated patients compared with a cohort of 410 patients with PMF who were treated with standard therapy at their center in the most recent 10-year period.47 Reasons reported for treatment discontinuation included disease progression or loss or lack of response (40%), toxicity with or without disease progression, or lack of response (34%). MDACC undertook a similar analysis, comparing the long-term outcomes of their patients with a historical control cohort of 310 patients culled from 3 databases who would have met eligibility for the study.48 OS was significantly improved in ruxolitinib-treated patients compared with historical controls adjusted for International Prognostic Scoring System risk group (HR = 0.58, P = .005).48 The 1-, 2-, and 3-year survival rates in high-risk patients treated with ruxolitinib were superior to the historical control group (95%, 83%, 63% vs 81%, 58%, and 35%, respectively; HR = 0.50, P = .006),46 and a nonsignificant trend in survival was observed between intermediate-2-risk ruxolitinib-treated and historical control patients.48 The discontinuation rates at 1, 2, and 3 years were 24%, 36%, and 46%, respectively, at MDACC vs 51%, 72%, and 89% for the Mayo Clinic and the mean total daily dose of ruxolitinib at MDACC was higher than at Mayo Clinic and was similar to the 31 mg total daily dose on the COMFORT-1 trial.47,48 Given the similarity in institutional baseline patient characteristics and caveats related to historical comparisons, the reported survival differences may be partly explained by dose duration and intensity, although additional factors likely contribute.47-49
Return of symptoms after hold of JAK inhibitor therapy
Interruption of ruxolitinib typically results in return of symptoms to baseline levels in approximately 1 week, a phenomenon that may be observed with other JAK inhibitors. After drug discontinuation, no drug-specific pattern of adverse events comprising a “withdrawal syndrome” unique to ruxolitinib or other JAK inhibitors has emerged. However, patients may develop worsening of their performance status, usually referable to signs and symptoms of MF, at a pace and severity that is variable and not predictable. Among the 51 patients treated by the Mayo Clinic in the phase 1/2 ruxolitinib study, 5 (11%) patients developed rapid return of symptoms, including painful enlargement of the spleen and hemodynamic decompensation.47 Tapering of the JAK inhibitor dose and/or use of corticosteroids may be a useful strategy in some patients.
Back to the bench: understanding disease resistance
In MF, clinical resistance to JAK inhibition can manifest as worsening blood counts (which must be distinguished from drug-related cytopenias), increasing splenomegaly, return of MF-related symptoms, and/or transformation to AML. Loss of response or disease progression50 may occur despite using higher doses of JAK inhibitors or in the setting of recurrent dose holds/reductions for hematologic or nonhematologic toxicity. Small increases in spleen size are usually clinically irrelevant if unaccompanied by worsening symptoms or blood counts. Consensus definitions of clinical resistance to JAK inhibitor therapy are now being formulated to guide physician management of patients. Although patients with “intolerance” or “resistance” to ruxolitinib are being evaluated in trials of alternate JAK inhibitors, the protocols do not use uniform criteria to define these terms.
“Disease persistence” is a phrase that has been used to describe the lack of in-depth responses or loss of clinical improvement on JAK inhibitor therapy. Thus far, sequencing of the JAK2 gene in small numbers of patients has not uncovered acquired resistance mutations. However, an vitro screen of libraries of mutagenized JAK2 V617F cDNA identified mutations in the drug-binding region that confer resistance to ruxolitinib.51 Five point mutations exhibited a 9- to 33-fold higher EC50 for ruxolitinib compared with JAK2 V617F and also exhibited cross-resistance to the JAK2 kinase inhibitors AZD1480, TG101348, lestaurtinib (CEP-701), and momelotinib.51 The “gatekeeper” mutation (M929I) in JAK2 V617F affected only ruxolitinib. In additional preclinical experiments, JAK2 V617F-expressing Ba/F3 cells exposed to ruxolitinib led to development of drug-resistant subclones that carried a novel JAK2 variant missing amino acids 76-820.52 The transcript results in the N-terminal FERM domain being directly fused to the kinase domain of JAK2 (FERM-JAK2). The FERM-JAK2 variant alters the JAK2 kinase domain structure, leads to direct, cytokine-receptor–independent STAT5 activation, and generates an accelerated, PV-like phenotype in mice. This short-form variant of JAK2 has not yet been reported in patients on JAK inhibitor therapy.
Levine et al found that cell lines and granulocytes from patients chronically treated with ruxolitinib displayed sustained phosphorylation of JAK2 that was related to an increased association between phosphorylated JAK2 and JAK family members JAK1 and TYK2.53 In addition, increased JAK2 mRNA expression and increased stability of the JAK2 protein contributed to persistence and facilitated heterodimerization between JAK family members. Withdrawal of JAK2 inhibitor resulted in resensitization to different JAK inhibitors and loss of association of JAK2 with JAK1/TYK2. There is an Hsp90 inhibitor that degrades JAK2 and a type II JAK inhibitor that also binds inactive JAK2-inhibited persistent cells, suggesting that alternative therapies could bypass the persistence phenomenon.53 However, in the clinical setting, the use of syncopated schedules of JAK inhibitors to resensitize patients to drug is impractical given the rapid return of disease symptoms upon treatment interruption.
JAK inhibitor therapy in PV and ET
The on-target effect of anemia and thrombocytopenia observed with most JAK inhibitors in MF is a hematologic profile that is exploitable in patients with PV and ET. The ongoing RESPONSE trial is evaluating ruxolitinib in PV and ET patients who are intolerant or refractory to hydroxyurea.54 In PV patients, ruxolitinib eliminated phlebotomy requirements and, in the majority of patients, normalized the WBC and platelet counts, reduced splenomegaly, and improved pruritus, night sweats, and bone pain. Among ET patients, 49% normalized platelet counts and 13/14 subjects with baseline platelet counts > 1000 × 109/L experienced a > 50% reduction.54 In PV and ET, the effects of JAK inhibitors on morbidity end points such as incidence of thrombosis or bleeding remain untested. In contrast to intermediate- or high-risk MF, where adjudicating effects on survival may be feasible, natural history studies in PV and ET are less tenable because of the long life expectancy of patients.
Potential uses of JAK inhibitors in other MPNs and myelodysplastic syndrome/MPNs
Therapeutic application of JAK inhibitors is being explored in the less common MPNs that exhibit low-frequency V617F mutations55 (or evidence of constitutive JAK-STAT pathway activation) such as chronic eosinophilic leukemia, systemic mastocytosis, chronic neutrophilic leukemia (CNL) and myelodysplastic syndrome/MPN overlap neoplasms such as chronic myelomonocytic leukemia, atypical CML, and refractory anemia with ring sideroblasts and thrombocytosis. The JAK2 pathway mediates antiapoptosis signals in response to the eosinophilopoietic cytokines IL-5 and GM-CSF, as well as the oncogenic tyrosine kinase FIP1L1-PDGFRA, suggesting a potential role of JAK inhibitors in eosinophilic myeloid disorders.56-58 At the time of this writing, 2 patients with chronic eosinophilic leukemia and a PCM1-JAK2 fusion [t(8;9)(p22;p24)] treated with ruxolitinib achieved complete hematologic remissions and cytogenetic responses (1 complete and 1 major).59,60 These in-depth responses indicate that ruxolitinib (and, by implication, other JAK inhibitors) may be more effective when JAK2 activation is mediated by chromosomal rearrangement rather than by point mutation, and could translate into alteration of the natural history of these rare diseases. Recently, CSF3R (G-CSF receptor) mutations were identified in ∼ 60% of CNL and atypical CML patients.61 CSF3R membrane proximal mutations activate the JAK/STAT pathway and exhibit sensitivity to JAK inhibitors.61 Treatment of a CSF3R T618I-mutated CNL patient with ruxolitinib resulted in a marked decrease in WBC and absolute neutrophil counts and resolution of thrombocytopenia. Another rational use of JAK inhibition is in Chuvash polycythemia, in which VHL mutants (eg, homozygous R200W and H191D) demonstrate altered affinity for SOCS1 and fail to form a heterodimeric E3 ligase that targets phosphorylated JAK2 for ubiquitin-mediated destruction. The JAK2 inhibitor TG101209 reversed the Chuvash phenotype in VHL knock-in mice.62 Finally, ruxolitinib (25 mg BID) has been evaluated in AML: among 38 patients with various types of refractory/relapsed leukemia, 3 responses (2 complete remissions and 1 complete remission with insufficient blood count recovery) were achieved among a subset of 18 patients with post-MPN AML, suggesting modest antileukemic efficacy in this patient population.63
Summary
The unprecedented success of tyrosine kinase inhibitors in CML created biased expectations of similar clinical triumphs with the use of JAK inhibitors in MF. However, this cognitive association ignores basic differences in the biology of the 2 MPNs. Improvements in spleen size, symptom burden, and QOL represent clinically meaningful advances with these agents. However, extended follow-up is required to gauge the implications of the survival data with ruxolitinib. Given that current JAK inhibitors are challenged by their inability to achieve hematologic remissions and that reductions of BM fibrosis or V617F allele burden may occur in only a subset of patients and are generally modest in nature,30,34,64,65 combination strategies with novel agents and the development of mutant-specific JAK inhibitors are being pursued. Application of JAK inhibitors in earlier stage MF, before genetic complexity and clinical complications supervene, and incorporation of these agents in the pretransplantation setting also merit further study.
Acknowledgments
The author thanks the Charles and Ann Johnson Foundation for their support of the Stanford MPN Center.
Disclosures
Conflict-of-interest disclosure: The author has received research funding from Gilead, Sanofi, and Incyte and has consulted for Gilead, Sanofi, and Incyte. Off-label drug use: Ruxolitinib; SAR302503; CYT387; Pacritinib.
Correspondence
Jason Gotlib, MD, MS, Associate Professor of Medicine (Hematology), Stanford Cancer Institute, 875 Blake Wilbur Dr, Rm 2324, Stanford, CA 94305-5821; Phone: 650-725-0744; Fax: 650-724-5203; e-mail: jason.gotlib@stanford.edu.