Abstract
Myelofibrosis (MF), including primary MF, postpolycythemia vera MF, and postessential thrombocythemia MF, is a clonal stem cell disorder characterized by BM fibrosis, extramedullary hematopoiesis, and a variable propensity to transform into acute leukemia. Allogeneic stem cell transplantation is the only known cure for MF, but its applicability is limited by the advanced age of most patients and by comorbid conditions. In the past decade, there has been an explosion of information on the molecular-genetic features associated with these diseases, fueled recently by the discovery of the JAK2V617F mutation. The development of JAK inhibitors has represented a significant therapeutic advance for these diseases; however, their use in MF has not yet been associated with eradication or a significant suppression of the malignant clone. In this era, much remains to be understood about MF, but it is likely that the identification of key pathogenetic drivers of the disease, coupled with the availability of novel molecularly targeted agents, will result in the discovery of new agents that significantly alter the natural history of the disease. This review focuses on recent and ongoing efforts in the development of novel agents in MF that go beyond the field of JAK inhibitors.
Introduction
The discovery of the JAK2V617F mutation in 2005 generated considerable excitement in the field of the Philadelphia-chromosome-negative (Ph−) myeloproliferative neoplasms (MPNs) and raised for the first time the question of whether these diseases may fall into the “one initiating mutation, one target” paradigm that has revolutionized the treatment of chronic myeloid leukemia. In the past few years, we have witnessed the clinical development of JAK-ATP mimetic inhibitors for the treatment of myelofibrosis (MF; used in this review to encompass primary MF, postpolycythemia vera MF, and post essential thrombocythemia MF) and have seen the therapeutic benefits for patients treated with these agents.1,2
It is clear, however, that there are limitations associated with the use of the currently approved JAK inhibitor ruxolitinib, as well as several other JAK inhibitors that are in clinical development. The clinical benefits to date with these agents have centered largely on effective palliation of symptoms and improvement in splenomegaly, although there is emerging evidence of a potential survival benefit with ruxolitinib.3 Significant areas of unmet need in the therapeutic arena include MF associated with significant cytopenias and/or transfusion dependency, accelerated or blast phase disease, and MF patients who progress on JAK inhibitors or who fail to achieve a response or experience clinical benefit with these agents.
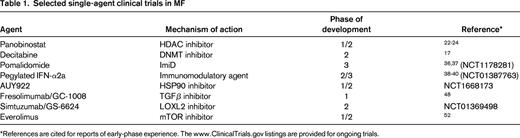
At the same time, the molecular complexity of the Ph− MPNs has become increasingly obvious (Figure 1) and these findings, along with the availability of novel agents for clinical investigation, is ushering in a new era in which novel targeted approaches can be pursued. Epigenetic therapies and approaches, immunomodulatory agents, agents that target the dysregulated cytokine milieu, and antifibrosis agents are being actively investigated (Table 1). In addition, there are various non-JAK kinase signal transduction inhibitors under investigation. This review focuses on the potential merits of and the experience thus far with these novel therapeutic approaches and highlights areas of ongoing and future investigation.

Gene mutations in PMF. Pie chart illustrates the molecular heterogeneity of primary MF (PMF) based on mutations in JAK2, ASXL1, TET2, SRSF2, DNMT3A, MPL, EZH2, CBL, IDH1, and IDH2. Asterisks depict mutations contributing to epigenetic dysregulation in PMF and/or occurring in epigenetic modifiers. Overlapping mutations (co-occurrence of 2 or more mutations in patients with PMF) are not depicted on this chart so the percentages add up to more than 100%. Chart is created from data derived from mutational analysis of all 10 markers in a cohort of 483 patients with PMF published by Vannucchi et al.7
Gene mutations in PMF. Pie chart illustrates the molecular heterogeneity of primary MF (PMF) based on mutations in JAK2, ASXL1, TET2, SRSF2, DNMT3A, MPL, EZH2, CBL, IDH1, and IDH2. Asterisks depict mutations contributing to epigenetic dysregulation in PMF and/or occurring in epigenetic modifiers. Overlapping mutations (co-occurrence of 2 or more mutations in patients with PMF) are not depicted on this chart so the percentages add up to more than 100%. Chart is created from data derived from mutational analysis of all 10 markers in a cohort of 483 patients with PMF published by Vannucchi et al.7
Epigenetic targets and therapies in MF
Epigenetic changes have generally been used to refer to heritable changes in gene expression that occur without a change in coding sequence. Epigenetic changes fall broadly into 2 major categories: changes in DNA methylation and changes in histone modifications. For example, recruitment of histone deacetylases (HDACs) and promoter DNA hypermethylation are 2 pathways of epigenetic silencing that are linked and are implicated in the transcriptional dysregulation underlying a variety of myeloid neoplasms. Unlike gene deletions, which lead to irreversible loss of function, transcriptional dysregulation by histone deacetylation or DNA methylation can potentially be reversed by chromatin-remodeling agents such as HDAC and DNA methyltransferase (DNMT) inhibitors, restoring tumor cells to a more transcriptionally normal state.4
For many years, there was a paucity of information on the scope of epigenetic changes in MF, and most of the initial studies published focused on analysis of the methylation status of individual genes of interest. Some of these studies provided evidence that epigenetic mechanisms silence genes that inhibit cellular proliferation in MF, such as the SOCS genes, which negatively regulate JAK/STAT signaling,5 or genes such as CXCR4,6 which regulates homing of stem cells to the BM. Perhaps one of the most compelling reasons to evoke epigenetic dysregulation as a central aspect of MF pathogenesis is the fact that, within the past few years, mutations in several genes that encode for proteins with a critical role in modifying the epigenome have been described (Figure 1).7 In addition, both wild-type and mutant JAK2 kinases have been demonstrated to participate in posttranslational modification of specific histone residues.8,9
Recently, MF has also been shown to have a distinct methylation signature (compared with normal controls and other Ph− MPNs) that consists of both aberrantly hypomethylated and hypermethylated loci.10 Pathways that were targeted by hypomethylation included genes affecting cell signaling, hematopoiesis, and immunologic pathways, whereas those that were targeted by hypermethylation included genes involved in inflammatory responses.
Overall, these findings raise the question of the clinical relevance of using chromatin-modifying agents or epigenetic modulators in MF. The potential promise of this approach has been suggested by the preclinical experience with these agents. For example, the use of an HDAC inhibitor in combination with a DNMT inhibitor in vitro led to a reduction in the numbers of MF progenitor CD34+ cells independently of the JAK2 mutational status. This is in contrast to the effects of these agents on normal primitive hematopoietic progenitor (CD34+) cells, where an expansion of these cells has been observed. The reduction in MF CD34+ cells was associated with up-regulation of CXCR4 transcript levels. In a NOD/SCID mouse model, these effects were associated with correction of the abnormal stem cell trafficking associated with this disease, resulting in homing of the cells to the BM, rather than to the spleen.11
HDAC inhibitors as single agents have been demonstrated in vitro to cause a down-modulation of JAK2V617F protein expression in JAK2 mutant cell lines and primary patient samples, but had no effect on the wild-type JAK2 protein.12 There was no effect on JAK2 mutant and wild-type transcript levels, suggesting that this effect was at the posttranscriptional level. It is likely that these inhibitory effects on the JAK2V617F protein were mediated at least in part through inhibition of HDAC6 and acetylation of HSP90,13 with resultant degradation of the JAK2 mutant protein (Figure 2). Both wild-type and mutant JAK2 proteins are known clients of HSP90, and the mutant protein may be more sensitive to degradation by the ubiquitin proteosome system in the context of disruption of the chaperone function of HSP90.14

Putative mechanism of action of HDAC inhibitors in MF. HSP90 is known to associate with HDAC6. JAK2 proteins (both wild-type and mutant proteins) are known clients of HSP90, which stabilizes these proteins. The use of a HDAC inhibitor leads to inhibition of HDAC6, with subsequent acetylation of HSP90 and resultant targeting of JAK2 proteins for proteosomal degradation. This mechanism may contribute at least in part to the down-modulation of the JAK2V617F protein observed with the use of HDAC inhibitors.
Putative mechanism of action of HDAC inhibitors in MF. HSP90 is known to associate with HDAC6. JAK2 proteins (both wild-type and mutant proteins) are known clients of HSP90, which stabilizes these proteins. The use of a HDAC inhibitor leads to inhibition of HDAC6, with subsequent acetylation of HSP90 and resultant targeting of JAK2 proteins for proteosomal degradation. This mechanism may contribute at least in part to the down-modulation of the JAK2V617F protein observed with the use of HDAC inhibitors.
Clinical investigation of DNMT inhibitors in MF
The clinical investigation of epigenetic modulators in MF thus far has been limited to a few small, early phase trials involving DNMT or HDAC inhibitors. The clinical efficacy reported in the majority of published reports has been modest. In a phase 2 study of the DNMT inhibitor 5-azacytidine administered on a 7-day schedule in 34 patients with MF, the overall response rate according to International Working Group (IWG) criteria was 24%, including 1 partial response. The majority of responses were clinical improvement in spleen size.15 A similar smaller study was conducted by the Mayo Clinic with the same agent in 10 patients. In the latter study, 5-azacytidine was administered on a 5-day schedule. No responses were observed. Only 2 patients in that study received more than 3 cycles.16 The most common reasons for treatment discontinuation were disease progression and toxicity. Grade 3/4 adverse toxicities observed were mainly hematologic in nature and included neutropenia and thrombocytopenia.
The DNMT inhibitor 5-aza-2′-deoxyxytidine (decitabine) has also been investigated in MF. In a preliminary report of a multicenter phase 2 trial in which low-dose decitabine was administered subcutaneously on a 10-day schedule, responses including improvement in anemia and thrombocytopenia were observed in 7 of 19 evaluable patients. This was associated with a sustained decline in circulating CD34+ cells in responding patients. Toxicity was largely hematologic; there were few nonhematologic side effects.17 There have also been published case reports of clinical benefit associated with decitabine use in MF.18
There is emerging evidence to suggest that DNMT inhibitors may be active in the treatment of Ph− MPNs that have progressed to acute myeloid leukemia or myelodysplastic syndrome (MDS).19,20 This is a subgroup of patients with a very dismal outcome, typified by an extremely short survival with conventional treatment approaches including intensive chemotherapeutic regimens.21 The Groupe Francophone des Myelodysplasies (GFM) has reported on the use of azacitidine in 54 patients with Ph− MPNs who had progressed to acute myeloid leukemia or MDS and reported a 52% overall response rate including 24% complete responses and 11% partial responses. Median response duration was 9 months, suggesting the need for additional alternative or consolidative approaches to produce further improvements in outcome.20
Clinical investigation of HDAC inhibitors in MF
HDAC inhibitors have also been investigated in clinical trials in MF. In a phase 1 dose-escalation study of panobinostat in 18 patients with MF,22 3 of 5 evaluable patients who received more than 6 cycles of therapy had an improvement in spleen volume and/or anemia. Reversible thrombocytopenia was the dose-limiting toxicity. Interestingly, one of these patients was reported to have achieved a near complete remission with significant reduction in BM fibrosis and resolution of splenomegaly and leukoerythoblastosis, effects that are unusual for nontransplantation therapy in this disease. The recommended phase 2 dose in this trial was 25 mg three times weekly, which was significantly lower than the doses used in other early phase trials conducted with this agent. The higher doses used in the latter trials were23,24 associated with significant fatigue, diarrhea, and thrombocytopenia requiring dose reductions and leading to early treatment discontinuation.23,24 Other HDAC inhibitors that have been investigated in MF with demonstrable but modest activity include givinostat25 and pracinostat.26 Interestingly, the responses observed were independent of the JAK2 mutational status, suggesting that additional work is necessary to identify the predictors of clinical activity given the pleiotropic effects of these agents and the mutational complexity of MF.
Combination approaches involving epigenetic modulators in MF
The clinical trials conducted to date with DNMT and HDAC inhibitors give some overall signal of modest efficacy. The experience with this class of agents in other myeloid neoplasms such as MDS would suggest that repeated cycles are necessary for optimum clinical activity. Ongoing challenges include the fact that optimum doses and schedules in MF are not yet defined and that MF patients may be more susceptible to myelosuppression with these agents, which may be dose limiting. There are ongoing efforts to determine optimal combinations, potentially using lower doses that may synergize with other agents and confer improvement in patient outcomes.
There is preclinical evidence in vitro and in murine models to suggest several potentially rational combinations. These include combination of an HDAC inhibitor with a DNMT inhibitor to simultaneously effect 2 pathways of epigenetic silencing in MF.11 In addition, because the antitumor activity of HDAC inhibitors in MF is likely due at least in part to inhibition of HDAC6, with subsequent acetylation of HSP90 and targeting of oncogenic JAK proteins for degradation by the ubiquitin-proteosome system,14,24,27 rational combinations of HDAC inhibitors plus JAK inhibitors could also be explored. This approach is supported by preclinical studies demonstrating synergy in vitro in JAK2V617F mutant cell lines and in vivo in murine models when HDAC inhibitors are combined with JAK inhibitors.14,28 There are already clinical trials in progress (Table 2) investigating some of the aforementioned combinations.
Additional work is necessary to determine whether other components of the epigenetic regulation machinery, such as histone methyltransferases, histone demethylases, histone acetyltransferases, and sirtuins, are potential therapeutic targets in MF. Recent preclinical studies suggest that the effects of mutant proteins such as IDH1 and IDH2, which contribute to epigenetic dysregulation in myeloid neoplasia, can be abrogated by specific inhibitors of those proteins.29,30 The development of inhibitors targeted to mutated components of the epigenetic machinery has the potential to transform the epigenetic therapeutic landscape in myeloid neoplasms including MF.
Immunomodulatory agents
Thalidomide analogs
Immunomodulatory drugs (ImiDs) are characterized by their ability to inhibit lipopolysaccharide-induced inflammatory cytokines such as TNFα, in addition to costimulation of T cells and augmentation of cytotoxic T- and NK-cell effector functions. The effects of these agents are pleiotropic and include antiangiogenic effects. More recently, epigenetic effects have also been demonstrated.31
Response rates with thalidomide-based regimens when reassessed recently by IWG criteria were in the 22% range for anemia and 8% for splenomegaly, with a median duration of 8.5 months.32 Peripheral neuropathy, constipation, and myelosuppression were the most notable toxicities. Twenty-two percent of patients overall failed to complete 3 cycles of therapy and the thalidomide-prednisone regimen was the most tolerable of the 3 thalidomide-based regimens evaluated.32 In a multicenter phase 2 study of lenalidomide and prednisone in MF, response rate by IWG criteria was 23%; most of these were clinical improvement in anemia, with myelosuppression being the most significant toxicity encountered.33 Significant activity has been reported in individual cases of MF associated with the 5q− cytogenetic aberration, which is a relatively rare subset of MF.34,35
Pomalidomide is the newest thalidomide analog to be investigated in MF based on the premise that ImiDs have measurable clinical activity in MF. In a phase 2, multicenter, randomized, double-blind, adaptive design study of pomalidomide in MF,36 4 treatment arms were evaluated including pomalidomide 2 mg daily plus placebo, pomalidomide 2 mg daily plus prednisone, pomalidomide 0.5 mg daily plus prednisone, and prednisone plus placebo. Prednisone was administered on a tapering schedule for 3 cycles starting at 30 mg/d in the first cycle. The IWG anemia response rates in the 4 treatment arms were 23%, 16%, 36%, and 19%, respectively. Median duration of response was 7.5 months. Pomalidomide was well tolerated in that trial, with relatively minimal myelosuppression observed (in contrast to prior experience with lenalidomide), and there were no significant reports of peripheral neuropathy (in contrast to prior experience with thalidomide). Overall, the results suggested that the 0.5 mg dose was at least equivalent with regard to efficacy and potentially less toxic. A subsequent report of the long-term outcome of 94 patients enrolled in 2 consecutive pomalidomide trials conducted at a single institution revealed an IWG anemia response rate of 27%, with a median duration of response of 16 months. The absence of a JAK2V617F mutation in the context of circulating blasts ≥ 5% or palpable splenomegaly ≥ 10 cm was negatively correlated with the achievement of a response.37
The results from these early phase trials provided a rationale for further investigation of pomalidomide in MF. Accrual has been completed for an ongoing placebo-controlled phase 3 trial of pomalidomide in transfusion-dependent MF (Table 1), and the results of this trial are eagerly awaited. Given the demonstrable activity of thalidomide analogs in improving anemia in MF, there are combination trials under way of ImiDs with other agents, including JAK inhibitors, in an effort to optimize the clinical activity of this class of drugs in MF (Table 2).
IFN
Multiple mechanisms have been proposed for IFN-α activity in myeloid neoplasia, including effects on immunomodulatory cells such as T cells and NK cells, induction of proapototic genes, suppression of proliferation of hematopoietic progenitor cells, and inhibition of angiogenesis.38 IFN-α at conventional doses in established MF is associated with significant myelosuppression and nonhematologic toxicities such as fatigue that have limited its use in MF. With the availability of more tolerable pegylated preparations and the demonstration that pegylated IFN can induce molecular responses in polycythemia vera, there has been a recent interest in investigating IFN in MF, particularly in MF patients in whom there is still a relative preservation of hematopoietic function.39,40 The clinical experience with IFN has recently been reviewed in detail.38 Larger prospective randomized trials (Table 1) will be required to validate the activity of this agent in early PMF, particularly with regard to the controversy surrounding the potential for reversal of BM histomorphologic changes with this agent.
Heat shock protein 90 inhibitors
Heat shock protein 90 (HSP90) serves as a molecular chaperone, resulting in the stabilization of several oncoproteins, so this molecule is often regarded as a facilitator of oncogenic addiction in various malignancies.41 HSP90 inhibition results in the targeting of oncogenic client proteins for proteosomal degradation, so HSP90 has emerged as a rational therapeutic target in various neoplasms. Both wild-type JAK2 and JAK2V617F have been demonstrated to be client proteins of HSP90.42 In addition, in vitro treatment of MPN cell lines and primary patient samples with HSP90 inhibitors resulted in degradation of JAK2 protein, inhibition of JAK/STAT signaling, and inhibition of cell growth.42,43
In JAK2 mutant murine models of PV and ET, in addition to the above effects, HSP90 inhibition led to normalization of peripheral blood counts, decline of the JAK2V617F allele burden, and improved survival in mice at a dose that did not affect JAK2 in normal tissues or cause significant toxicity.42 Interestingly, heterodimeric JAK/STAT activation (heterodimerization of JAK2 with JAK1 or TYK2) has been implicated as a mechanism of persistent disease in the face of JAK2 inhibitor therapy and cell lines that harbored this mechanism of activated JAK2 persistence retained their sensitivity to HSP90 inhibitors.44 In addition, cells engineered to express JAK2 kinase domain mutations that conferred resistance to JAK-ATP mimetic inhibitors also retained their sensitivity to HSP90 inhibitors.45 These findings provide a compelling rationale for the clinical investigation of HSP90 inhibitors in MF.
Antifibrosis agents
Allogeneic stem cell transplantation is the only therapeutic modality to date that has been demonstrated conclusively to lead to a reversal of fibrosis in MF. The pathogenetic mechanisms underlying the development of fibrosis in MF are poorly understood. Although MF has been established to be a clonal disorder of pluripotent stem cell origin, the fibroblasts have been shown to be polyclonal and are functionally similar to normal fibroblasts. The BM findings in MF include increases in the numbers of stromal cells and in levels of extracellular matrix proteins, angiogenesis, and osteosclerosis.
Monoclonal antibodies
A current pathogenetic hypothesis with regard to the genesis of fibrosis in MF is that clonal megakaryocytes secrete fibrogenic and angiogenic cytokines including TGFβ and MMP-9.46,47 A common theme emerging from several murine models of MF is that there is a significant association between increased numbers of megakaryocytes and the development of an extracellular matrix typical of MF, and these findings lend credence to the hypothesis that megakaryocytes play a central role in fibrogenesis.47 The availability of novel agents targeting specific pathways and cytokines implicated in fibrogenesis has led to a resurgence in interest with regard to the clinical investigation of antifibrosis agents in MF.
For example, a monoclonal inhibitor of TGFβ, fresolimumab (GC-1008), was recently investigated in a phase 1 trial in MF48 and was found to be associated with a significant decline in TGFβ1 levels in both evaluable patients enrolled in that trial. The trial was terminated early due to drug supply issues after enrollment of just 3 patients. The potential merit of targeting TGFβ in MF is supported by a recent study identifying abnormal genetic signatures in TGFβ1 signaling in the spleen and BM from the GATA-1low mouse, a murine model of MF.49 Inhibition of TGFβ1 signaling in this mouse model normalized this aberrant gene expression signature, restored hematopoiesis and megakaryocyte development, and reduced fibrosis, neovascularization, and osteogenesis in the BM.49
A different monoclonal antibody, simtuzumab (GS-6624), is being studied in a phase 2 clinical trial in MF. Simtuzumab is a monoclonal antibody against lysyl oxidase (LOX)-like protein 2. LOX is a copper-dependent enzyme that initiates the covalent cross-linking of collagen or elastin and was shown to be abundant in the GATA-1low mouse, which is characterized by significant MF and high levels of low-ploidy megakaryocytes. Inhibition of LOX enzymatic activity led to a significant improvement of the fibrotic phenotype, leading to the speculation that LOX is a potential therapeutic target in diseases such as MF.47,50
Aurora kinase inhibitors
Another potential target of the megakaryocyte-fibrosis axis are aurora kinases. In acute megakaryocytic leukemia, a disease with a dismal prognosis characterized by expansion of immature megakaryocytes and profound BM fibrosis, aurora kinase A was identified as a mediator of polyploidization of malignant megakaryocytes. Aurora kinase A inhibition in acute megakaryocytic leukemia led to apoptosis of malignant megakaryocytes and induced polyploidization and expression of mature megakaryocyte markers.51 Given the prominent role of clonal megakaryocytes in MF, these findings have led to the speculation that aurora kinase A could be a valid therapeutic target in MF, and there are ongoing preclinical studies to validate these assertions.
Non-JAK kinase signal transduction inhibitors
There several signaling intermediates downstream of the JAK/STAT signaling pathway (Figure 3) that constitute rational therapeutic targets in MF. The PI3K/AKT/mammalian target of rapamycin (mTOR) pathway has been shown to be dysregulated in MPNs and AKT activation has been found to be critical for JAK2V617F-mediated cellular transformation. AKT and mTOR inhibitors have been associated with growth inhibition of primary MPN cells and cell lines in vitro. These findings have led to the clinical investigation of mTOR inhibitors in MF.52 In a phase 1/2 study of everolimus in MF, 20% of patients achieved a rapid/sustained reduction in splenomegaly of > 50% and more than two-thirds of patients enrolled experienced complete resolution of constitutional symptoms and improvement in pruritus. A minority of patients (15%-25%) experienced improvement in anemia and thrombocytopenia. Although known targets of mTOR such as phosphor-p70S6K were identified as potential biomarkers of response to the agent, there was no significant effect on MF-related biomarkers such as JAK2V617F allelic burden, circulating CD34+ cells, or cytokine levels. The drug was well tolerated, with grade 1-2 stomatitis being the most common toxicity.

Novel agents targeting pathways downstream of the JAK/STAT signaling pathway in MF. Activated JAK2 signals through and activates downstream signaling intermediates such as STAT5, RAS/MAPK, and PI3K/AKT/mTOR pathways, leading to effects on proliferation and survival of MPN cells. JAK proteins can be down-modulated by the use of HSP90 inhibitors or HDAC inhibitors, which lead to targeting of both wild-type and mutant proteins for degradation by the proteosomal system. PI3K/AKT inhibitors, mTOR inhibitors, MEK inhibitors, and STAT inhibitors can inhibit the respective signaling intermediates downstream of JAK/STAT pathway. DNMT inhibitors can potentially reverse epigenetic silencing of various genes including the SOCS genes, which are negative regulators of the JAK/STAT signaling pathway.
Novel agents targeting pathways downstream of the JAK/STAT signaling pathway in MF. Activated JAK2 signals through and activates downstream signaling intermediates such as STAT5, RAS/MAPK, and PI3K/AKT/mTOR pathways, leading to effects on proliferation and survival of MPN cells. JAK proteins can be down-modulated by the use of HSP90 inhibitors or HDAC inhibitors, which lead to targeting of both wild-type and mutant proteins for degradation by the proteosomal system. PI3K/AKT inhibitors, mTOR inhibitors, MEK inhibitors, and STAT inhibitors can inhibit the respective signaling intermediates downstream of JAK/STAT pathway. DNMT inhibitors can potentially reverse epigenetic silencing of various genes including the SOCS genes, which are negative regulators of the JAK/STAT signaling pathway.
Potent inhibitors of the PI3K/AKT pathway are entering into clinical trials in MF. Combinations of JAK inhibitors and PI3K inhibitors may also be reasonable (Table 2) based on emerging preclinical evidence that suggests synergy.
How I treat MF in the JAK kinase inhibitor era
Given the variability in outcome associated with MF, a comprehensive assessment including risk stratification according to contemporary prognostic scoring models such as the Dynamic International Prognostic Scoring System (DIPSS)53,54 or DIPSS plus55 is essential to help guide treatment recommendations. I use a watch and wait approach in asymptomatic lower (low- and intermediate-1) risk patients, whereas those with higher (intermediate-2 and high-) risk disease require intervention (Figure 4). Given the emerging data for improved tolerance with reduced intensity conditioning transplantation regimens, early referral for allogeneic stem cell transplantation should be considered in transplantation-eligible patients.56 I recommend JAK inhibitor therapy for symptomatic patients with MF, particularly in the absence of significant cytopenias. In the presence of significant cytopenias or for patients with prior JAK inhibitor exposure, I enroll patients in a clinical trial whenever possible. For MF-accelerated or blast phase disease, a particularly poor prognostic subset of patients,21 immediate referral for allogeneic stem cell transplantation in transplantation-eligible patients should be strongly considered. In these instances, I recommend hypomethylating agent therapy17,19,20 whenever feasible or cytarabine-based induction approaches for cytoreduction and/or conversion to a second chronic phase of the disease before proceeding with transplantation.
![Figure 4. How I treat MF in the JAK inhibitor era. The DIPSS can be used for risk stratification at any time point during the course of MF. Prognostic variables are each assigned a score of 1 point, except for anemia, which is assigned a score of 2 points. These variables include: age > 65 years, anemia (defined as hemoglobin < 10 g/dL), WBC count > 25 × 109/L, circulating blasts ≥ 1%, and constitutional systems. The recommended treatment algorithm according to risk stratum categories (low, intermediate 1 [Int-1]; intermediate 2 [Int-2], and high) is summarized in the figure. Early referral for allogeneic stem cell transplantation and/or enrollment in clinical trials is recommended for symptomatic or higher risk patients with significant cytopenias (hemoglobin < 10 g/dL, platelet count < 100 K/μL) and/or prior JAK inhibitor exposure. In the absence of an appropriate clinical trial, ImiD agents, androgenic steroids, or erythropoietin-stimulating agents can be considered, particularly for anemic patients.](/view-large/figure/6513830/bep0011302910004.jpeg)
How I treat MF in the JAK inhibitor era. The DIPSS can be used for risk stratification at any time point during the course of MF. Prognostic variables are each assigned a score of 1 point, except for anemia, which is assigned a score of 2 points. These variables include: age > 65 years, anemia (defined as hemoglobin < 10 g/dL), WBC count > 25 × 109/L, circulating blasts ≥ 1%, and constitutional systems. The recommended treatment algorithm according to risk stratum categories (low, intermediate 1 [Int-1]; intermediate 2 [Int-2], and high) is summarized in the figure. Early referral for allogeneic stem cell transplantation and/or enrollment in clinical trials is recommended for symptomatic or higher risk patients with significant cytopenias (hemoglobin < 10 g/dL, platelet count < 100 K/μL) and/or prior JAK inhibitor exposure. In the absence of an appropriate clinical trial, ImiD agents, androgenic steroids, or erythropoietin-stimulating agents can be considered, particularly for anemic patients.
How I treat MF in the JAK inhibitor era. The DIPSS can be used for risk stratification at any time point during the course of MF. Prognostic variables are each assigned a score of 1 point, except for anemia, which is assigned a score of 2 points. These variables include: age > 65 years, anemia (defined as hemoglobin < 10 g/dL), WBC count > 25 × 109/L, circulating blasts ≥ 1%, and constitutional systems. The recommended treatment algorithm according to risk stratum categories (low, intermediate 1 [Int-1]; intermediate 2 [Int-2], and high) is summarized in the figure. Early referral for allogeneic stem cell transplantation and/or enrollment in clinical trials is recommended for symptomatic or higher risk patients with significant cytopenias (hemoglobin < 10 g/dL, platelet count < 100 K/μL) and/or prior JAK inhibitor exposure. In the absence of an appropriate clinical trial, ImiD agents, androgenic steroids, or erythropoietin-stimulating agents can be considered, particularly for anemic patients.
Conclusion
It has been many decades since Damashek's original description of the MPNs, and MF has remained a largely incurable disease. The discovery of the JAK2V617F mutation and the development of JAK inhibitors have been a significant step forward, and have fueled the resurgence of interest in this group of diseases. Recent biologic insights underscore the molecular complexity of this group of diseases and it will be critical to decipher which genetic and epigenetic aberrations are the key drivers. This knowledge, along with an increasing plethora of new agents that target pathways of potential pathogenetic importance in MF, is likely to bring us closer to an agent or agents that will significantly change the natural history of this disease.
Disclosures
Conflict-of-interest disclosure: The author is on the board of directors or an advisory committee for Suneisis Pharmaceuticals, Spectrum Pharmaceuticals, Sanofi-Aventis, and Incyte and has received research funding from Topotarget and Eisai. Off-label drug use: None disclosed.
Correspondence
Olatoyosi Odenike, Department of Medicine and Comprehensive Cancer Center, The University of Chicago, 5841 South Maryland Ave, Chicago, IL 60637; Phone: 773-702-3354; Fax: 773-834-0188; e-mail: todenike@medicine.bsd.uchicago.edu.