Abstract
Our understanding of the genetic basis of the Philadelphia chromosome-negative myeloproliferative neoplasms (MPNs) has moved forward at a staggering pace over the last decade. With the discoveries of underlying mutations in JAK2, MPL, and, most recently, calreticulin (CALR), that together account for ∼90% of patients with MPNs, these conditions are now among the best characterized of hematological malignancies. While JAK-STAT pathway activation has been shown to be central to the pathogenesis of the MPN phenotype, the mechanism by which mutant CALR alters cellular function to result in myeloid proliferation remains unclear. Other mutations in several epigenetic modifiers, such as ASXL1, DNMT3a, TET2, EZH2, IDH1, and IDH2, as well as in genes involved in mRNA splicing, such as SF3B1 and U2AF2, have also been described in recent years in patients with MPNs, and evidence is emerging as to how these may be contributing to disease biology. From a therapeutic perspective, the discovery of aberrations in JAK2 has rapidly translated into the successful clinical use of JAK inhibitors in MPNs. Mutant calreticulin has the potential to be a tumor-specific therapeutic target because the mutations generate a novel protein C-terminus. In this chapter, we detail the genomic alterations that underlie MPNs, with a focus on the recent discovery of mutations in CALR, and explore the clinical and biological relevance of the altered genomic landscape in MPNs.
Learning Objectives
To review the spectrum of genetic abnormalities in MPNs and their biological significance
To recognize the clonal complexity and clonal evolution within MPNs and how this may relate to clinical phenotype
To recognize the clinical relevance of testing for specific genetic mutations for both diagnosis and prognosis of MPNs
Somatic mutations that activate JAK-STAT signaling
In 1951, William Dameshek described prescient observations that polycythemia vera (PV), essential thrombocythemia (ET), and myelofibrosis (MF) were a set of closely interrelated disorders.1 X-chromosome-inactivation studies in females subsequently revealed that myeloproliferative neoplasms (MPNs) were clonally derived neoplasms2 and, in 2005, landmark findings showed that somatically acquired mutations in the gene JAK2 were found in the majority of patients with MPNs.3-6 The point mutation V617F in exon 14 of JAK2, together with rare insertions and deletions in exon 12,7 account for the majority of patients with PV, and JAK2V617F is also found in 60% of patients with ET or MF. How the JAK2 mutation results in the development of any 1 of 3 different MPNs has been the subject of great interest to researchers and has been reviewed previously (reviewed elsewhere). Overall, evidence is accumulating that JAK2-mutated PV or ET may be better viewed as a continuum with factors such as levels of iron stores and erythropoietin, sex, genetic modifiers, and levels of JAK2 homozygosity or allele burden helping to shape the phenotype seen in patients between ET and PV.8-10
Multiple clinical studies have shown that JAK2-unmutated MPNs (encompassing ∼1/2 of patients with ET or MF) represent distinct MPN subgroups with differences in clinical features at presentation and clinical outcome.8,10,11 In 2006, our understanding of the pathogenesis of these cases progressed with the discovery that 3%–5% of JAK2-unmutated ET cases and 8%–11% of JAK2-unmutated MF cases carry mutations in exon 10 of the thrombopoietin receptor gene MPL (Figure 1).12,13 The most common mutations affect codon 515 (MPLW515L and MPLW515K), but other mutations are also occasionally seen (eg, MPLS505N)14,15 and homozygosity for MPL mutations is associated with increased BM fibrosis.16 Clinically, MPL-mutated MPN patients are, on average, older and have a lower hemoglobin than their MPL-unmutated counterparts.14,17,18 In ET, MPL mutations are also associated with higher platelet counts and reduced BM cellularity with reduced erythropoiesis compared with JAK2-mutated ET.14 However, no differences in clinical outcome have thus far been demonstrated for this small subgroup of MPN patients in either ET or MF.17,19

Frequency of JAK2, CALR, and MPL mutations in PV, ET, and MF.JAK2 mutations are found in 97% of patients with PV and in 50%–60% of those with ET and MF. CALR mutations are the next most frequent genetic aberration, affecting 1/3 of patients with ET or MF. MPL mutations are found in 3%–10% of ET or MF patients. JAK2, MPL, and CALR mutations are mutually exclusive in the majority of patients. Ten to fifteen percent of ET and MF cases have no common underlying genetic marker.
Frequency of JAK2, CALR, and MPL mutations in PV, ET, and MF.JAK2 mutations are found in 97% of patients with PV and in 50%–60% of those with ET and MF. CALR mutations are the next most frequent genetic aberration, affecting 1/3 of patients with ET or MF. MPL mutations are found in 3%–10% of ET or MF patients. JAK2, MPL, and CALR mutations are mutually exclusive in the majority of patients. Ten to fifteen percent of ET and MF cases have no common underlying genetic marker.
Studies into the mechanisms by which abnormal JAK2 and MPL lead to myeloid proliferation have established a central role for overactive JAK-STAT signaling and cytokine-independent growth as the underlying pathogenesis of MPNs. Mutations in JAK2 and MPL are both gain-of-function mutations and result in cytokine-independent growth in cell lines (Ba/F3) with activation of downstream JAK-STAT signaling.3,12,15 Although different murine models of JAK2V617F exhibit disease phenotypes ranging from ET to PV and MF,20 murine models of MPL mutations develop a disease marked by thrombocytosis with features of ET and MF.21 These differences in disease phenotype recapitulated in cell lines and animal models appear to mimic the clinical spectrum of MPNs with which these different mutations are associated, and are probably reflective of the differing expression patterns of JAK2 and MPL in hematopoietic progenitors.
Infrequently, alternative targets in the JAK-STAT pathway are affected by somatic mutations in MPNs. SH2B3 (LNK) is an inhibitor of erythropoietin and thrombopoietin signaling and mutations result in loss of function and increased JAK-STAT signaling.22 CBL mutations abrogate their ubiquitin ligase activity, resulting in reduced degradation of tyrosine kinases and prolonged activation of intracellular signaling.23 LNK mutations are found in 2%–6% of all MPN subtypes and CBL mutations are more frequent in MF (5%–10%), secondary acute myeloid leukemia (AML), and other myeloid malignancies.24
Mutations in CALR in the majority of JAK2/MPL-unmutated ET and MF
Despite the discoveries of mutations in JAK2 and MPL, 40% of ET and MF patients still lacked a common genetic basis, and diagnosing these patients in the clinical setting remained a challenge, requiring BM biopsies and exclusion of reactive causes. It had been hypothesized by researchers that alternative candidates may be mutated that overactivate cytokine signaling in a manner analogous to JAK2 mutations, however, no such abnormalities were identified. Next-generation sequencing technologies were being harnessed by many groups to sequence the whole exomes or genomes of MPN patients. In 2012, Hou et al reported single cell sequencing of a JAK2-unmutated MPN and were able to confirm a monoclonal origin, but the somatic mutations identified were not found to be recurrently mutated in other patients.25 In December 2013, the groups of Tony Green/Peter Campbell and Robert Kralovics reported results of 2 independent studies that identified highly recurrent somatic mutations in the gene calreticulin (CALR) in JAK2/MPL-unmutated ET and MF (Figures 1, 2).26,27 Klampfl et al performed whole-exome sequencing in 6 patients with JAK2/MPL-unmutated MF and targeted resequencing of greater than 1000 MPNs and Nangalia et al published whole-exome sequencing results of 151 patients across PV, ET, and MF, with targeted resequencing of a 3000 patient follow-up series.26,27

Somatic mutations in CALR and predicted consequences. (A) Terminal exon (exon 9) of the gene CALR. The 2 most common mutations, found in 85% of cases, are a 52-bp deletion (Type 1; L367fs*46) or a 5-bp insertion of TTGTC (Type 2; K385fs*47) shown above the exon in blue. Rarer mutations comprise other deletions, insertions, or complex mutations and are shown below the exon. Grey bars represent deleted DNA and black letters show inserted bases. (B) Wild-type amino acid peptide sequence of the CALR C-terminus and the predicted sequence of the 2 most common mutations. All mutations lead to a 1-bp shift to the reading frame, resulting in the generation of a common novel terminal peptide sequence (shaded in blue). (C) Predicted effects on protein structure. Wild-type CALR on the left has a C-domain rich in calcium-binding sites and a KDEL endoplasmic reticulum retention motif, both of which are predicted to be lost in mutant CALR, as shown on the right.
Somatic mutations in CALR and predicted consequences. (A) Terminal exon (exon 9) of the gene CALR. The 2 most common mutations, found in 85% of cases, are a 52-bp deletion (Type 1; L367fs*46) or a 5-bp insertion of TTGTC (Type 2; K385fs*47) shown above the exon in blue. Rarer mutations comprise other deletions, insertions, or complex mutations and are shown below the exon. Grey bars represent deleted DNA and black letters show inserted bases. (B) Wild-type amino acid peptide sequence of the CALR C-terminus and the predicted sequence of the 2 most common mutations. All mutations lead to a 1-bp shift to the reading frame, resulting in the generation of a common novel terminal peptide sequence (shaded in blue). (C) Predicted effects on protein structure. Wild-type CALR on the left has a C-domain rich in calcium-binding sites and a KDEL endoplasmic reticulum retention motif, both of which are predicted to be lost in mutant CALR, as shown on the right.
CALR mutations were found in 60%–88% of patients with ET and MF who were negative for JAK2 and MPL mutations, comprising 1/4 to 1/3 of patients with ET and MF. Identification of these mutations was not a trivial matter because they were easily missed by standard bioinformatic algorithms for several reasons. DNA capture and sequencing coverage over the mutated region of CALR was low (median 10× DNA reads per patient sample) compared with equivalent regions of JAK2, which were well captured and sequenced to a high depth (median >100× DNA reads per patient sample).26 In addition, the mutated region of CALR is repetitive in nature and thus prone to misalignment of DNA reads. Last, large deletions and insertions are less readily identifiable by commonly used algorithms for identification of such changes after next-generation sequencing. Nevertheless, standard algorithms identified the mutations in a few patients and subsequent extensive manual curation of the sequence data revealed the true frequency of CALR mutations in JAK/MPL-unmutated MPNs.26 CALR mutations are also infrequently found in myelodysplastic syndrome (MDS), refractory anemia with ringed sideroblasts and thrombocytosis (RARS-T), and, very rarely, in cases of atypical chronic myeloid leukaemia or chronic myelomonocytic leukaemia (CMML), but have not been found in other myeloid or lymphoid malignancies, solid tumors, or healthy controls.26,27 Overall, these results demonstrate a striking specificity for JAK2-unmutated ET and MF that is highly reminiscent of the disease spectrum associated with mutations in MPL.
CALR mutations are all insertions or deletions in the DNA sequence of the terminal exon (exon 9) of the gene and the 2 most common mutations accounting for ∼85% of mutated cases are either a 52-bp deletion (Type 1; c.1099_1150del; L367fs*46; 44%–53% of cases) or a 5-bp insertion (Type 2; c.1154_1155insTTGTC; K385fs*47; 32%–42% of cases) (Figure 2). The remaining 15% of cases comprise other insertions, deletions, or a combination of both, and these mutations are often unique to, or found in, a few patients each.28 Regardless of the mutation, the number of nucleic acids inserted or deleted is such that the reading frame of the mRNA is shifted by exactly 1 base pair, leading to the coding of a novel amino acid peptide sequence distal to the site of the mutation that generates a novel C-terminus (Figure 2). This pattern of mutations is similar to alterations in nucleophosmin 1 (NPM1) in AML, which also involves insertions or deletions in the DNA sequence that result in a common reading frame change affecting the terminal part of the protein amino acid sequence.29 In the case of mutant NPM1, the neomorphic protein lacks a nuclear localization signal that is present in the wild-type protein.29
Calreticulin is a highly conserved luminal ER chaperone protein that ensures the proper folding of newly synthesized glycoproteins, but it has also been implicated in several other roles both within and outside of the ER, including calcium homeostasis, immunogenic cell death, proliferation, and apoptosis.30,31 Mutant calreticulin's novel protein C-terminus differs from the wild-type protein in 2 ways. First, a Golgi-to-ER retention signaling motif (KDEL) responsible for retrieving and retaining chaperone proteins back to the ER is lost in the mutant protein. Initial studies of whether mutant CALR is mislocalized out of the ER have had mixed results, but it appears that mutant CALR remains largely within the ER.26,27 Second, the largely negatively charged and acidic C-terminus of the wild-type protein is replaced by a basic and positively charged novel peptide sequence (Figure 2), and this has been predicted to alter the calcium-binding capacity of the protein.32 How these changes to the C-terminus result in myeloid proliferation with a specificity for abnormal megakaryopoiesis remain obscure. Preliminary evidence suggests that mutant calreticulin may also lead to overactivation of JAK-STAT signaling: transcriptional studies have shown that activated JAK2 signaling is seen in all MPN patients, including those with CALR mutations33 and, in addition, activated STAT5 has been found in cell lines transfected with mutant but not wild-type CALR.27 However, the mechanism by which this signaling perturbation may occur remains unclear. Mutant calreticulin may alter the structure, trafficking, or activation status of the thrombopoietin receptor MPL with subsequent JAK-STAT activation or it may lead to activation of megakaryopoiesis through alternative signaling cascades such as altered calcium signaling.
CALR mutations have been shown to be acquired at the level of the hematopoietic stem cell (HSC) and clonal characterization of MPN samples has shown that mutated CALR is present in the earliest clone, which is consistent with it being an initiating event in these malignancies.26,27 Mutant CALR has since been shown to be highly expressed in, and largely restricted to, the megakaryocyte lineage in primary patient bone marrrow trephines immunostained with an antibody selective to mutant CALR's novel C-terminus.34 Interestingly, this study also found that wild-type calreticulin had a megakaryocyte-restricted expression pattern, which may explain why the mutant form is associated with MPNs characterized predominantly by abnormal megakaryopoiesis.34
Clinically, CALR-mutated ET is associated with a higher platelet count, lower hemoglobin, and lower leucocyte count than JAK2-mutated counterparts, and patients are younger and more frequently male.10,35 Patients also have a reduced incidence of thrombosis that remains significant after correction for age. This may be important for future risk stratification of patients, although no differences in survival have thus far been demonstrated for the different mutation subgroups in ET.10,35 CALR-mutated MF patients have a significantly better overall survival compared with those harboring either JAK2V617F and MPLW515 mutations.27 This observation has been confirmed to be independent of age and the DIPSS-plus prognostic score.11 Interestingly, this latter study also showed that the majority of triple-negative MF patients (ie, those without mutations in JAK2, CALR, or MPL), carried mutations in other genes, or harbored cytogenetic abnormalities, confirming the clonal origin of this subset of MF.
Early evidence is emerging as to the differential effects of the various mutations in CALR. In MF, Type 1/L367fs*46 CALR mutations are found more frequently than Type 2/K385fs*47 mutations11,26,27 and may also be associated with a shortened survival.36 In ET, there appears to be no difference in clinical course or outcome between the 2 most common CALR mutations, although Type 2/K385fs*47 CALR mutations are associated with a higher platelet count at diagnosis.26,27,37
Mutations in epigenetic regulators
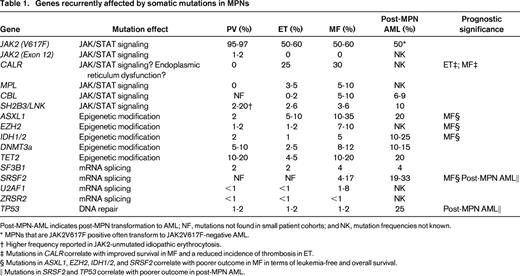
JAK2, MPL, and CALR mutations together provide a genetic marker for the majority of MPNs (99% of PV and 85% of ET and MF). However, several other genes involved in DNA methylation (TET2, DNMT3A, IDH1/2)38-41 or chromatin structure (EZH2, ASXL1)42,43 have also been shown to be mutated in MPNs (Figure 3, Table 1). Mutations in epigenetic regulators are not specific to MPNs and are associated with many myeloid malignancies, but within MPNs, these mutations are often found in patients who also carry JAK2, MPL, or CALR mutations, indicating a cooperation between these 2 classes of mutations in MPN pathogenesis.

Other genes frequently mutated in MPNs affect epigenetic regulation or mRNA splicing. Genes affected by loss-of-function mutations (DNMT3a, TET2, ASXL1, and EZH2) are shown in blue and genes affected by gain-of-function mutations (IDH1/2) and mutations affecting spliceosome function are shown in pink. 2-HG indicates 2-hydroxyglutarate; αKG, alpha-ketoglutarate; K27, methylated lysine 27 tail of histone H3. CpG sites are shown in white (unmethylated), red (5-methylcytosine, 5-mc), or grey (5-hydroxymethylcytosine, 5-hmc), depending on their methylation status.
Other genes frequently mutated in MPNs affect epigenetic regulation or mRNA splicing. Genes affected by loss-of-function mutations (DNMT3a, TET2, ASXL1, and EZH2) are shown in blue and genes affected by gain-of-function mutations (IDH1/2) and mutations affecting spliceosome function are shown in pink. 2-HG indicates 2-hydroxyglutarate; αKG, alpha-ketoglutarate; K27, methylated lysine 27 tail of histone H3. CpG sites are shown in white (unmethylated), red (5-methylcytosine, 5-mc), or grey (5-hydroxymethylcytosine, 5-hmc), depending on their methylation status.
DNA methylation at the 5 position of cytosines (5-methylcytosine, 5-mc) in CpG dinucleotides is a major epigenetic mechanism by which gene expression is regulated during normal development, and altered in neoplasia. DNA methytransferases (DNMTs) 3a and 3b carry out de novo methylation, unlike DNMT1, which maintains existing methylation patterns after cell division, and the TET (Ten-eleven translocation methylcytosine dioxygenase) family of proteins undertake initial steps in demethylating DNA by converting 5-mc to 5-hydroxymethylcytosine (5-hmc), as well as other intermediates. Mutations in TET2 were first identified in 2009 in patients with MDS, AML, and MPNs by interrogating regions of acquired rearrangements on chromosome 4q24 present in patients.38,44 Loss-of-function mutations in TET2 are found in 5%–17% of MPNs and have been shown to result in reduced levels of 5-hmc.24,45 TET2 plays an important role in normal myelopoeisis and murine models with disrupted TET2 display an expansion of the HSC compartment, myelomonocytic proliferation, and features resembling CMML.46 In MPNs, TET2 mutations have been associated with an increased risk of leukemic transformation and shorter survival. However, other studies have not found such clinical associations47,48 and larger studies are required to establish the true clinical significance of aberrant TET2 in MPNs. Interestingly, TET2 mutations have been found in healthy elderly females who display clonal hematopoiesis in the absence of any blood count parameter abnormalities.49 This finding, in conjunction with data from murine models, suggests that TET2 mutations may confer a clonal advantage at the level of the HSC rather than driving the overproduction of erythroid and/or megakaryocyte cells that is characteristic of MPNs. Data from studies assessing the biological effects of combinations of JAK2 and TET2 mutations, as well as the significance of the order in which they are acquired, are awaited.
DNMT3a, a de novo DNMT, is mutated in 4%–15% of MPNs, less frequently than in AML, where they were initially discovered.24,50 DNMT3a R882H is the most common mutation and it has recently been demonstrated that this variant exerts a dominant-negative effect on wild-type DNMT3a and results in myeloid proliferation with thrombocytosis in a murine model.51,52 In addition, serial transplantation experiments using bone marrow from DNMT3a-knockout mice have demonstrated that loss of DNMT3a results in marked HSC expansion.53 Recently, patients with AML were found to have acquired DNMT3a mutations in HSC populations before the acquisition of additional mutations found in AML blasts (eg, mutations in NPM1).54 These DNMT3a-mutated preleukemic HSCs displayed a multilineage repopulation advantage over wild-type HSCs, confirming that acquisition of mutant DNMT3a provides a clonal advantage.54 In MPNs, DNMT3a mutations can occur before or after the acquisition of mutations in JAK2,48 but, clinically, studies to date have not demonstrated any associations between DNMT3a mutations and alterations to MPN phenotype or outcome.
Posttranslational modification of histones and regulation of chromatin structure is another major mechanism by which gene expression is regulated. The polycomb repressive complex 2 (PRC2) functions as a transcriptional repressor by catalyzing dimethylation and trimethylation of lysine 27 at histone H3 (H3K27me2/3), which is associated with gene-silencing and chromatin compaction. ASXL1 (Addition of Sex Combs Like 1) has been shown to be an important mediator of PRC2 function and also interacts with BAP1, a nuclear deubiquitinating enzyme.55 DNA deletions, insertions, and nonsense mutations in ASXL1 are found in ∼20% of MDS cases and more frequently in CMML. In MPNs, mutations are found in 2%–10% of PV and ET cases, but are found in ∼1/4 of MF (both primary MF and post-PV or ET), where they are associated with more severe anemia and an inferior survival that is independent of the DIPSS-Plus score.24,26,56,57 ASXL1 has been shown to be critical for normal myelopoiesis and disruption of the gene in a murine model results in anemia, leukopenia, and morphological dysplasia resembling myelodysplasia.58 Interestingly, this study also found significant extramedullary hemopoiesis, splenomegaly, and expansion of the HSC compartment with a block to erythroid differentiation, features that are characteristic of ASXL1-mutated MF.58
The enzymatic component of the PRC2 complex, H3K27 methyltransferase EZH2, is also mutated in MPNs at a frequency of 3%–13%. Mutations are most frequent in MF, where they are associated with an increased risk of leukemic transformation and an inferior survival.59 As with ASXL1, mutations in EZH2 are predicted to be loss-of-function mutations, and deletion of the EZH2 homolog in murine models results in an MDS/MPN phenotype that is exacerbated by concurrent deletion of TET2.60 Other PRC2 components, such as SUZ12, EED, and JARID2, are also rarely found to be mutated in patients with MPNs.24,61 Overall, impairment of PRC2 function appears to be a very important pathogenic mechanism that promotes malignant myelopoiesis. Interestingly, the loss-of-function mutation spectrum affecting PRC2 components in myeloid malignancies is very different from that seen in lymphomas. In germinal center-derived diffuse large B cell lymphomas and follicular lymphomas, recurrent gain of function mutations are found in EZH2 at Y641 that are predicted to result in increased H3K27 methylation, suggesting that PRC2 gain-of-function may, conversely, contribute to malignant lymphomagenesis.62
IDH1 and IDH2 are enzymes that catalyze the conversion of isocitrate to alpha-ketoglutarate. Mutations are present in ∼20% of blast phase MPNs, where they confer an inferior overall survival and are present in 1%–4% of chronic phase MPNs.63,64 The specific mutations result in neomorphic enzymatic activity with excess production of 2-hydroxyglutarate.65 This has been shown to prevent histone demethylation and to block hematopoietic differentiation,66 and mutant IDH1/2 also impairs the function of alpha-ketoglutarate-dependent proteins such as TET2.67
Mutations in genes involved in mRNA splicing
Genes that alter mRNA splicing (SF3B1, U2AF1, SRSF2) are enriched in dysplastic myeloid disorders. SF3B1 mutations are frequent in RARS, whereas SRSF2 mutations are highly recurrent in CMML.68,69 In MPNs, mutations in this group of genes affect ∼5% of patients, with SF3B1 mutations most frequent in RARS-T.27 Recent data from MF patients suggest that the presence of SRSF2 mutations confer a poor prognosis, with reduced overall and leukemia-free survival.57
Genomic landscape of chronic-phase and accelerated-phase MPNs
Classical cytogenetic karyotype analysis has always shown that chromosomal translocations and large-scale chromosomal aberrations are infrequent in MPNs, with only occasional patients harboring, for example, del20q, del13q, trisomy 8, and del9p. This suggested a relatively simple and unaltered genomic landscape in MPNs. However, more recently, microarray technology that captures chromosomal copy number information at much greater resolution than conventional cytogenetic analysis has identified greater complexity in these diseases, with subtle chromosomal aberrations and acquired losses of heterozygosity demonstrated in up to 2/3 of MPN patients.70 The most common chromosomal abnormality in MPNs is loss of heterozygosity by uniparental disomy (UPD) for 9p, which is found in association with the JAK2 mutation and results in homozygosity for mutant JAK2. Other regions of UPD in the genome are also seen in the context of underlying mutations within affected regions, such as MPL mutations and 1p UPD. 19p UPD and homozygosity for mutant CALR also occurs for both Type 1/L367fs*46 and Type 2/K385fs*47 mutations,71 but this is an infrequent event and the clinical consequences are unknown. Some cytogenetic abnormalities, such as del20q, have not been shown to be associated with a genetic mutation within the abnormal chromosomal region and recent data have shed mechanistic insights into how this may be contributing to MPN biology by showing that the resultant loss of expression of imprinted genes L3MBTL and SGK2 within the commonly deleted region on 20q dysregulates erythropoiesis and megakaryopoiesis.72
Next-generation sequencing technologies now allow us to interrogate whole exomes or genomes at single-base resolution. A comprehensive exome-sequencing study of 151 MPNs has shown that DNA mutations show a predominance of C → T transitions as found in other myeloid malignancies and solid tumors, and reflects the spontaneous deamination of 5-methylcytosines.26 PV and ET samples harbor, on average, 6-7 somatic mutations per patient, and MF samples have twice as many mutations, which is consistent with it being a more advanced phase of the disease.26 The number of mutations in MPNs is, in fact, similar to other hematological malignancies (eg de novo AML, ALL, and CLL; median 8-13 somatic mutations/sample), but much lower than epithelial cancers (median 40-70 somatic mutations/tumor).73 Overall, an increasing number of somatic mutations are seen at disease progression and have been shown to be a poor prognostic marker in MPNs associated with reduced leukemia-free and overall survival.48 Acquisition of specific mutations are associated with disease progression to MF, such as ASXL1, EZH2, IDH1/2, and SRSF2, as discussed previously.
At transformation to post MPN-AML, cytogenetic abnormalities are often complex. TP53 mutations have been shown to be present in 1/4 of patients at leukemic transformation and the majority of patients harbor biallelic TP53 loss either through acquisition of independent mutations on both TP53 alleles or monoallelic mutation followed by 17p UPD48,74 In patients with secondary AML that do not carry TP53 mutations, a substantial proportion have been found to have amplification of 1q, which contains MDM4, a potent p53 inhibitor. Together, p53 loss at leukemic transformation of MPNs may be seen in 1/2 of patients.74 IDH1/2 and SRSF2 are also seen more frequently in AMLs transformed from preceding MPNs.57,64
Clonal complexity in MPNs
From a biological point of view, MPNs provide a window into early tumorigenesis that can be interrogated at a clonal level. Unexpected clonal complexity in MPNs was first suggested by findings that JAK2-mutated MPNs often transformed to JAK2-unmutated AML and supported the existence of independent or pre-JAK2 clones.75 Identification of further mutations in MPNs and delineation of clonal structures in patient samples have revealed significant clonal complexity with the existence of multiple mutant subclones at any one time.48 Although, initially, it was shown that mutations in TET2 predate JAK2 mutations, it is now clear that mutations in epigenetic regulators can occur before or after the acquisition of JAK2V617F, as well as in separate clones.48 What the biological and phenotypic consequences are of these differing patterns of orders of acquisition is currently unknown and is a major question for cancer in general. A model depicting the disease initiation, clonal heterogeneity, and evolution of MPNs is shown in Figure 4.

Model depicting the development and evolution of MPNs. Monoclonal expansion of HSCs can occur in healthy individuals and often precedes acquisition of mutations that drive the phenotype of MPNs. JAK2, MPL, and CALR mutations and other genetic or patient factors influence the MPN phenotype seen in patients (PV, red-shaded cells; ET, blue-shaded cells). MPNs are clonally heterogeneous, with multiple mutant subclones existing within neoplasms. Subclonal expansion can be associated with acceleration of disease, the phenotype and prognosis of which is determined by the nature of additional somatic mutations or chromosomal aberrations acquired. Transformation to AML can occur from early or late subclones.
Model depicting the development and evolution of MPNs. Monoclonal expansion of HSCs can occur in healthy individuals and often precedes acquisition of mutations that drive the phenotype of MPNs. JAK2, MPL, and CALR mutations and other genetic or patient factors influence the MPN phenotype seen in patients (PV, red-shaded cells; ET, blue-shaded cells). MPNs are clonally heterogeneous, with multiple mutant subclones existing within neoplasms. Subclonal expansion can be associated with acceleration of disease, the phenotype and prognosis of which is determined by the nature of additional somatic mutations or chromosomal aberrations acquired. Transformation to AML can occur from early or late subclones.
Germline predisposition to MPNs
Familial MPNs, in which there is clustering of at least 2 MPN cases within a family pedigree, has been reported to affect ∼5% of apparently sporadic cases.76 The majority of affected family members also have somatic mutations in JAK2 and MPL, as seen in the sporadic MPN setting, and it appears that family members are at an increased predisposition to acquiring these somatic mutations than the general population. Many of the family members affected by MPNs and were JAK2/MPL unmutated have now been shown to harbor somatic CALR mutations.77,78 Recently, inherited mutations in RBBP6 were found in one such pedigree, which appeared to account for the germline predisposition to acquiring MPNs in family members.79 Familial MPN families are not to be confused with families affected by hereditary erythrocytosis or thrombocytosis. In these latter families, nonclonal myeloproliferation is caused by several different germline mutations in genes known to affect erythropoietin or thrombopoietin signaling (for review, see Jones and Cross80 ) or in genes in which the mechanism of myeloproliferation remains less clear (eg. mutations in gelsolin).81
In terms of predisposition to MPNs in the general population, the strongest inherited factor remains the JAK2 constitutional 46/1 or “GGCC” haplotype, which has been estimated to account for 50% of the population-attributable risk of developing an MPN.80 Recently, another germline sequence variant, rs2736100_C, in the gene TERT has been reported to be associated with both MPNs and higher blood count indices in healthy individuals in an Icelandic cohort and awaits confirmation in other populations.82
Clinical importance of the mutational landscape in MPNs
The World Health Organization (WHO) diagnostic criteria were revised in 2008 to encorporate molecular markers such as JAK2V617F, JAK2 Exon 12, and MPL mutations as new major diagnostic criteria for MPNs.83 Screening for CALR mutations is now also part of the diagnostic workup of suspected ET or MF in many centers internationally and further revisions to WHO criteria are expected.
Several studies have undertaken comprehensive mutational analysis of large cohorts, including prospective cohorts from clinical trials, to characterize the prognostic significance of the multitude of mutations found in MPNs. In MF, the presence of mutations in any one of ASXL1, SRSF2, IDH1/2, or EZH2 has been shown to be associated with a poorer survival and increased leukemic transformation.57 Specifically, the number of these “high-molecular-risk” (HMR) mutations was inversely correlated with median survival in MF independently of current prognostic scoring systems (2.6, 7, and 12.3 years for cases with ≥2, 1, or no HMR mutations).84 Analysis of the prospective COMFORT-II cohort, which compared ruxolitinib with best available therapy in patients with MF, showed that ruxolitinib improved survival for such HMR patients, which is the first indication that treatment choices may influence the natural history of MF with HMR mutations.85 Similar studies in PV and ET are awaited. Clinical studies of retrospective cohorts have shown that CALR mutations are associated with a better prognosis and survival in MF and reduced thrombosis in ET,10,11,35 and this may be relevant for future risk stratification of patients. Validation of these findings in prospective clinical trial cohorts is awaited.
The emergence of a multitude of epigenetic targets in MPNs and myeloid malignancies has led to the testing of many novel agents as part of clinical trials. Whether these drugs are particularly effective in MPN subgroups that harbor specific mutations remains to be determined. Mutant CALR in ET and MF certainly holds promise as a tumor-specific therapeutic target given the neomorphic C-terminus of the mutant protein and its selective expression in megakaryocytes.34
Concluding remarks and future perspectives
Our understanding of the genomic landscape of MPNs has leapt forward over the last decade. JAK2 mutation testing allows diagnosis of 99% of patients with PV, and combined testing for JAK2, MPL, and CALR mutations captures 85%–90% of patients with ET and MF. The causative genetic lesions in 10%–15% of ET and MF cases remains unknown at this stage and this subset may include patients with nonclonal myeloproliferation due to reactive causes, germline genetic aberrations, other myeloid disorders, or as-yet-unidentified somatic mutations within the exome or genome. We are beginning to understand the prognostic impact of coexisting mutations in epigenetic or spliceosome regulators in MPNs, but whether this should affect treatment decisions remains to be tested in future clinical trials. Discovery of mutations in JAK2 and epigenetic regulators has led to the successful use of JAK inhibitors in MF and ongoing trials with multiple other novel agents, and the recent identification of mutant CALR has the potential for the development of a tumor- and genotype-specific targeted therapy.
Disclosures
Conflict-of-interest disclosures: The authors declare no competing interests. Off-label drug use: None disclosed.
Correspondence
Tony R. Green, Department of Haematology, Cambridge Institute for Medical Research, University of Cambridge, Hills Road, Cambridge CB2 0XY, United Kingdom; Phone: +44-1223-336820; Fax: +44-1223-762670; e-mail: arg1000@cam.ac.uk.