Abstract
Anticoagulants currently used in clinical practice to treat or prevent thromboembolic disease are effective, but place patients at increased risk for serious bleeding because they interfere with plasma enzymes (thrombin and factor Xa) that are essential for hemostasis. In the past 10 years, work with genetically altered mice and studies in baboons and rabbits have demonstrated that the plasma contact proteases factor XI, factor XII, and prekallikrein contribute to the formation of occlusive thrombi despite having limited roles in hemostasis. In the case of factor XI, epidemiologic data from human populations indicate that elevated levels of this protein increase risk for stroke and venous thromboembolism and may also influence risk for myocardial infarction. These findings suggest that inhibiting contact activation may produce an antithrombotic effect without significantly compromising hemostasis. This chapter reviews strategies that are being developed for therapeutic targeting of factor XI and factor XII and their performances in preclinical and early human trials.
Learning Objective
To understand that the proteases of the plasma contact system play a limited role in hemostasis, but appear to make significant contributions to thrombosis; therefore, targeting of contact activation may produce an antithrombotic effect that is not accompanied by a drug-induced defect in hemostasis
Introduction
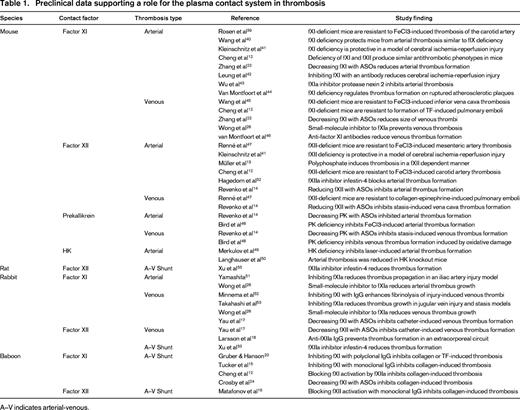
In the traditional coagulation cascade (Figure 1A), thrombin formation is initiated by a process called contact activation, which is triggered when plasma is exposed to certain types of surfaces (usually those with a negative charge).1,2 Contact activation involves reciprocal conversion of the protease precursors factor XII (fXII) and prekallikrein (PK) to their active forms (fXIIa and α-kallikrein) in the presence of the cofactor high-molecular-weight kininogen (HK). FXIIa then converts factor XI (fXI) to its active form (fXIa), setting off a series of enzymatic reactions that culminate in thrombin generation. Contact activation initiates clotting in the activated partial thromboplastin time (aPTT) assay used widely in clinical practice to assess the integrity of the blood coagulation mechanism. Therefore, plasmas lacking fXII, fXI, PK, or HK have very long aPTTs.2 Despite this, individuals deficient in fXII, PK, or HK do not have a demonstrable bleeding disorder even when challenged with surgery.1,2 Patients with fXI deficiency can bleed excessively when surgery or trauma involves certain tissues, but spontaneous bleeding is rare and symptoms are considerably less severe than with deficiency of factor IX (the substrate for fXIa in the coagulation cascade).2-4 A conclusion that can be drawn from the clinical observations is that clot formation at a wound site does not behave like the chain of reactions depicted in Figure 1A, in which the absence of any link would be expected to disable the entire mechanism. Work over the past 40 years has clarified the processes responsible for initiation and propagation of a clot at a site of injury. Our current understanding of the main reactions involved in thrombin generation at a wound site are summarized in Figure 1B.5 In this scheme, fXI serves a relatively small role in hemostasis, whereas the process of contact activation is no longer considered an integral component of the hemostasic mechanism. However, as discussed by Key and by van Montfoort and Meijers in separate chapters in this publication, data from human population studies, supported by numerous studies with animal models (summarized in Table 1), make a strong case for a role for fXI in thromboembolic disorders. Work with the animal models also suggests that fXII, PK, and HK contribute to thrombosis (Table 1). These exciting observations are the driving force behind efforts to develop strategies that target components of contact activation for therapeutic purposes.

Models of thrombin generation. (A) Contact activation-initiated thrombin generation. In the cascade/waterfall hypothesis of coagulation, thrombin generation is initiated by the process contact activation (gray oval). Contact activation involves reciprocal activation of the protease precursors fXII and PK on a surface (typically a negatively charged surface). HK serves as a cofactor for the reaction by facilitating PK binding to the surface. FXIIa then activates fXI, in a reaction that also requires HK, setting off the series of calcium-dependent proteolytic reactions that culminates in thrombin generation. (B) TF-initiated thrombin generation. In this more current scheme, thrombin generation is initiated by factor VIIa in plasma binding to TF, a membrane protein expressed on the surface of cells beneath the blood vessel endothelium. The factor VIIa/TF complex activates factor X to factor Xa and factor IX to factor IXa. Factor Xa converts prothrombin to thrombin in the presence of factor Va and factor IXa sustains the process by activating additional factor X in the presence of factor VIIIa. The reactions indicated by the black arrows form the core of the thrombin-generation mechanism in vertebrate animals. Mammals have fXIa, which provides another mechanism for fIX activation (red arrow). Although fXI is activated by fXIIa during contact activation, this reaction is not shown in this scheme because it does not appear to be required for hemostasis. FXI can be activated by thrombin generated early in the coagulation process (gray arrows), explaining the lack of a bleeding disorder in people lacking fXII. In (A) and (B), the precursors (zymogens) of trypsin-like enzymes are indicated in black lettering, with active forms indicated by a lowercase “a.” Protein cofactors are indicated by Roman numerals in yellow ovals.
Models of thrombin generation. (A) Contact activation-initiated thrombin generation. In the cascade/waterfall hypothesis of coagulation, thrombin generation is initiated by the process contact activation (gray oval). Contact activation involves reciprocal activation of the protease precursors fXII and PK on a surface (typically a negatively charged surface). HK serves as a cofactor for the reaction by facilitating PK binding to the surface. FXIIa then activates fXI, in a reaction that also requires HK, setting off the series of calcium-dependent proteolytic reactions that culminates in thrombin generation. (B) TF-initiated thrombin generation. In this more current scheme, thrombin generation is initiated by factor VIIa in plasma binding to TF, a membrane protein expressed on the surface of cells beneath the blood vessel endothelium. The factor VIIa/TF complex activates factor X to factor Xa and factor IX to factor IXa. Factor Xa converts prothrombin to thrombin in the presence of factor Va and factor IXa sustains the process by activating additional factor X in the presence of factor VIIIa. The reactions indicated by the black arrows form the core of the thrombin-generation mechanism in vertebrate animals. Mammals have fXIa, which provides another mechanism for fIX activation (red arrow). Although fXI is activated by fXIIa during contact activation, this reaction is not shown in this scheme because it does not appear to be required for hemostasis. FXI can be activated by thrombin generated early in the coagulation process (gray arrows), explaining the lack of a bleeding disorder in people lacking fXII. In (A) and (B), the precursors (zymogens) of trypsin-like enzymes are indicated in black lettering, with active forms indicated by a lowercase “a.” Protein cofactors are indicated by Roman numerals in yellow ovals.
Rationale for therapeutic targeting of contact proteases
The importance of the protease thrombin in thromboembolic disease has been established and it follows that inhibition of thrombin production or thrombin activity will limit or prevent thrombus formation and growth. For more than 60 years, 2 approaches have been used to manipulate thrombin to treat or to prevent thromboembolic disease. One approach is based on inhibiting the enzymatic activity of thrombin or factor Xa (the protease directly responsible for converting prothrombin to thrombin; Figure 1B). This can be achieved indirectly using unfractionated heparin, low-molecular-weight heparin, or heparin-related compounds to enhance protease inhibition by the plasma serine protease inhibitor antithrombin.6 More recently, small-molecule inhibitors that target the active sites of thrombin or factor Xa directly have been used toward the same end.7 An alternative approach involves reducing synthesis of functional prothrombin and factor X, the zymogen precursors of thrombin and factor Xa, respectively, by administration of vitamin K antagonists such as warfarin.8 The strategy of targeting thrombin and fXa activity or production to achieve an antithrombotic effect is based on the reasonable premise that thrombosis represents dysregulation of processes normally involved in hemostasis. Certainly, from the standpoint of efficacy, this strategy has an impressive track record. However, because of the importance of thrombin and factor Xa to hemostasis, patients on anticoagulation therapy are at increased risk for severe bleeding. In clinical trials, the newer oral direct thrombin and factor Xa inhibitors were associated with similar numbers of bleeding episodes compared with warfarin or low-molecular-weight heparin, with the exception of the factor Xa inhibitor apixaban, which appeared safer than warfarin in phase 3 trials. Therefore, life-threatening bleeding complications continue to be a problem, even with newer therapies, because they interfere with key components of a vital host-defense mechanism.
The normal hemostatic response to injury observed in humans lacking fXII, PK, or HK and the relatively mild bleeding associated with fXI deficiency led to the conclusion that the contact system plays, at most, a minor role in coagulation in mammals. However, this notion is being reconsidered because a growing body of evidence supports a substantive role for the contact factors in thrombus formation in a variety of pathologic conditions. As discussed by Key in this publication, higher plasma fXI levels in humans increase the risk for stroke9 and venous thromboembolism10 and may also affect risk for myocardial infarction.11 Consistent with this, as reviewed by van Montfoort and Meijers in this publication, mice lacking fXI, fXII, or PK have an impressive resistance to injury-induced arterial and venous thrombosis despite lacking an obvious hemostatic abnormality (Table 1).12-14 Intravascular thrombi are unstable in these mice, preventing clots from achieving a size sufficient to occlude a vessel. Data from ex vivo studies of human blood under flow support the impression that growth of an intraluminal thrombus requires fXIa and fXIIa activity15,16 either within the clot or on its surface. In this regard, intraluminal thrombi appear to differ from hemostatic clots that form within the walls of injured blood vessels. Therefore, contact activation appears to contribute to clot formation in vivo, but is only required for the growth of pathologic occlusive clots. If this hypothesis is correct, it raises the prospect that inhibiting contact activation or components of contact activation could produce an antithrombotic effect that would be associated with little or no antihemostatic (anticoagulant) effect. Based on the intriguing epidemiologic and preclinical data, a variety of strategies are under development for specifically targeting zymogen fXI and fXII and their active protease forms, fXIa and fXIIa, for therapeutic purposes. These approaches are reviewed in the following sections and are summarized in Table 2.
Inhibiting fXI and fXII with monoclonal antibodies in a primate thrombosis model
Although data from both human population studies and animal models support the premise that fXI contributes to thrombosis, the data for fXII present a more confusing picture. Specifically, although fXII clearly serves an important role in thrombus formation in a variety of rodent models12-14 and in rabbit models of artificial surface-induced thrombosis,17,18 epidemiologic data suggest that there is actually an inverse relationship between plasma fXII levels and risk of myocardial infarction and death from cardiac disease in humans.11,19 Although the reasons for this apparent discrepancy are not clear, the data raise concerns that fXII may not contribute to thrombus formation in the same manner, or to the same extent, in rodents and primates. This issue has been addressed recently by a series of studies examining the consequences of fXI and fXII inhibition in a baboon thrombosis model.
In 2003, Gruber and Hanson reported that olive baboons (Papio anubis) treated with a polyclonal antibody to human fXI were resistant to occlusive thrombus formation induced by insertion of Dacron- or tissue factor (TF)-coated Teflon grafts into the circulation.20 Several monoclonal antibodies that block different steps in the contact activation process have subsequently been tested in a version of this model that involves placing collagen-coated grafts into temporary femoral arteriovenous shunts.12,15,16 Accumulation of 111In-labeled platelets and 125I-labeled fibrin within and downstream of the graft can be followed to determine the effects of antibodies on the rate and extent of thrombus formation. The most impressive results to date were obtained with the antihuman fXI IgG O1A6 (also called aXIMAb).15 O1A6 binds to the third “apple” (A3) domain of fXI/fXIa (Figure 2). The A3 domain contains a binding site for the fXIa substrate factor IX (indicated in red in Figure 2),21 and O1A6 prevents factor IX from binding to this site. In baboons, a single 2 mg/kg subcutaneous dose of O1A6 reduced plasma fXI activity to nearly undetectable levels for 10 days. During this time, the concentration of plasma fXI antigen increased, indicating that O1A6 produced its effect by blocking fXI activity, not by inducing rapid fXI clearance from plasma. Platelet accumulation within collagen-coated grafts was markedly reduced in O1A6-treated animals compared with controls. Platelet adhesion to the collagen-coated surface was actually unaffected by the antibody, but subsequent platelet aggregation was blunted, preventing graft occlusion. Similar to what was observed in mice lacking fXI or fXII, platelet-rich aggregates were unstable in the presence of O1A6 and fragmented in the flowing blood. Thrombus instability may well have been due, at least partly, to a significant reduction in local thrombin generation because plasma levels of thrombin-antithrombin complex (TAT, a marker of thrombin generation) were reduced by >90% immediately downstream from the graft. Consistent with this, fibrin deposition in the graft was reduced by >80% compared with controls. O1A6 was considerably more effective at inhibiting thrombus growth than high-dose aspirin in this model and at least as effective as unfractionated and low-molecular-weight heparin. O1A6-treated baboons did not have any manifestations of abnormal hemostasis.

Human fXI. This schematic diagram shows the primary amino acid sequence, general domain structure, and the disulfide bonds (cysteine residues shown as the letter C in black circles) for a human fXI subunit. Each subunit is comprised of 4 apple domains (A1 through A4) and a C-terminal trypsin-like catalytic domain. The mature molecule in plasma is a dimer of 2 of these subunits connected through a hydrophobic interface on the A4 domains, with the cysteines at position 321 in each subunit forming an interchain-disulfide bond. Conversion of fXI to fXIa involves proteolytic cleavage of the bond after arginine 369 (R369, indicated by the black arrow). This cleavage can be produced by fXIIa or thrombin. Amino acids thought to be required for fIX binding to the fXIa A3 domain are indicated in red and residues that comprise the heparin binding site on the catalytic domain are shown in blue. The position of the active site serine residue (S557) is indicated in yellow. (Used with permission from McMullen et al.59 )
Human fXI. This schematic diagram shows the primary amino acid sequence, general domain structure, and the disulfide bonds (cysteine residues shown as the letter C in black circles) for a human fXI subunit. Each subunit is comprised of 4 apple domains (A1 through A4) and a C-terminal trypsin-like catalytic domain. The mature molecule in plasma is a dimer of 2 of these subunits connected through a hydrophobic interface on the A4 domains, with the cysteines at position 321 in each subunit forming an interchain-disulfide bond. Conversion of fXI to fXIa involves proteolytic cleavage of the bond after arginine 369 (R369, indicated by the black arrow). This cleavage can be produced by fXIIa or thrombin. Amino acids thought to be required for fIX binding to the fXIa A3 domain are indicated in red and residues that comprise the heparin binding site on the catalytic domain are shown in blue. The position of the active site serine residue (S557) is indicated in yellow. (Used with permission from McMullen et al.59 )
The monoclonal IgG 14E11 was raised in fXI-deficient mice and binds to the A2 domain (Figure 2) of fXI and fXIa from multiple species, including humans and baboons.12 In plasma, 14E11 effectively interferes with fXI activation by fXIIa, but not fXI activation by thrombin. It also has little effect on fXIa activation of factor IX. Therefore, 14E11 appears to specifically inhibit contact activation-mediated fXI activation. In baboons, 14E11 reduced fibrin deposition within grafts by ∼40%, but platelet accumulation was largely unchanged and the effect on TAT generation was more modest than with O1A6. However, the antibody significantly reduced platelet accumulation and thrombus growth downstream of the graft, consistent with a significant antithrombotic effect. Similar results to those obtained with 14E11 were reported earlier this year in the baboon model for the antihuman fXII antibody 15H8.16 15H8 is a strong inhibitor of activation of fXII to fXIIa, but has little inhibitory effect on fXIIa itself.
Taken as a whole, the data support a role for both fXI and fXII in thrombus formation in primates, but suggest that inhibiting fXIa may be a more effective antithrombotic strategy than inhibiting fXI activation by fXIIa or fXII activation. O1A6, 14E11, and 15H8 had comparable effects on the aPTTs of treated baboons. This demonstrates that it is not the degree of prolongation of the aPTT that is indicative of the antithrombotic effect as much as the step in the process that is being inhibited. The findings are consistent with the data for human populations, which make a stronger case for a role for fXI in thrombosis than for fXII. Perhaps there is a contribution to fXI activation through a fXIIa-independent mechanism during thrombosis in baboons, such as the thrombin-mediated feedback reaction represented by the gray arrows in Figure 1B. It is important to recognize, however, that the results of the baboon studies could be explained by differences in how effectively each antibody inhibits its specific target reaction and the results should not be interpreted as definitively demonstrating that fXI is a better antithrombotic target than is fXII.
Reducing plasma fXI levels with antisense oligonucleotides
The plasma levels of fXI, fXII, or PK can be significantly reduced in mice by subcutaneous administration of antisense-oligonucleotides (ASOs) specific for the mRNA of the protein of interest.14,22 Like their congenitally deficient counterparts, ASO-treated mice demonstrate a marked resistance to injury-induced arterial and venous thrombosis.14,22 Deoxyribonucleotide-based ASOs bind to a target mRNA through complementary base pairing,22 leading to selective mRNA degradation by cellular nucleases and, consequently, reduced synthesis of the protein encoded by the mRNA. ASOs administered by subcutaneous injection are taken up avidly by hepatocytes, facilitating targeting of proteins such as coagulation factors that are synthesized in the liver. ASOs complementary to mouse fXI mRNA have been prepared that are ∼20 nucleotides in length, with phosphorothionate incorporated into the backbone and 2′-O-methoxyethyl modifications in the 5 nucleotides at each end.22 Such “second-generation” ASOs have relatively long tissue elimination half-lives and can be administered at intervals of several days to a week. Administration of 50 mg/kg of an anti-fXI ASO subcutaneously twice a week for 3 weeks to adult mice resulted in >90% reduction of plasma fXI protein and activity. The treated mice are as resistant to arterial and venous thrombosis as mice treated with warfarin or heparin. A dose escalation study revealed that an antithrombotic effect becomes evident when the plasma fXI level is reduced to ≤20% of the normal level.
More recently, ASO technology has been studied in primates. A dose escalation trial (4-40 mg/kg/wk) of the anti-fXI ASO ISIS 416858 in cynomolgus monkeys produced a dose-dependent reduction in plasma fXI, with higher doses producing an 80% reduction after 4 weeks of treatment.23 As expected, the aPTT was moderately increased by this therapy, but the PT and platelet count remained normal and there was no evidence of a hemostatic deficit in response to tail amputation or gum or skin laceration. In the baboon arteriovenous shunt thrombosis model described in the previous section, reducing fXI by as little as 50% had some antithrombotic effect, with a greater effect occurring when the plasma level was reduced to ≤20% of normal,24 consistent with results from the studies with mice.22
Results of a phase 1 study examining the effects of the antihuman fXI ASO ISIS-FXIRx on healthy volunteers were reported at the annual meeting of the American Society of Hematology in 2011.25 Reductions of plasma fXI antigen and activity of ∼80% were consistently achieved with repeated 200 or 300 mg doses of ASO, with some individuals experiencing >95% reduction. The most common adverse effects were mild irritation and inflammation at ASO injection sites. There were no cases of excessive bleeding, significant hematologic or electrolyte abnormalities, or liver or kidney dysfunction. Salomon et al reported that patients with severe fXI deficiency requiring orthopedic procedures often experience little excessive bleeding, even in the absence of factor replacement.4 A multicenter phase 2 trial comparing ISIS-FXIRx with low-molecular-weight heparin for deep vein thrombosis prophylaxis in patients undergoing total knee arthroplasty is currently under way. This study will provide us with information on the effects of reduced fXI in a clinical situation associated with a high incidence of procedure-associated deep vein thrombosis and, as importantly, on the effects of fXI reduction on perioperative bleeding.
Targeting fXII to prevent thrombus formation on artificial surfaces
Although contact factors, fXI in particular, may contribute to venous and arterial thrombosis in humans, the factor VIIa/TF complex (Figure 1B) may be a more important trigger of thrombus formation in these disorders.5,26 However, there are clinical situations in which it seems reasonable to postulate that contact activation may be the predominant initiator of abnormal coagulation. The propensity for the contact system to become activated when blood interacts with artificial surfaces may trigger blood coagulation and inflammation during cardiopulmonary bypass or extracorporeal membrane oxygenation (ECMO) or in patients with ventricular assist devices used as bridges to heart transplantation or chronic indwelling catheters for venous access. Two studies published earlier this year make a strong case for a fXII-initiated process contributing to thrombosis induced by artificial surfaces. Yau et al17 studied the effects of ASO-induced factor VII, fXI, fXII, or HK reduction on thrombus formation induced by placement of polyurethane catheters into the jugular veins of rabbits. Reductions of ∼90% of the targeted protein were achieved with all ASOs. ASOs targeting fXII and fXI prolonged the time to thrombus formation more than 2-fold, whereas those reducing fVII and HK had little effect. The negative results with HK knockdown may suggest that the process is somewhat different from the contact activation that triggers clotting in the aPTT assay (which is highly dependent on HK). However, the small amount of residual HK in the animals in this study may have been sufficient to support classic contact activation.
Larsson et al18 recently demonstrated that a recombinant human antibody, designated 3F7, that targets the active site of fXIIa was as effective as heparin in preventing thrombus formation in rabbits connected to a pediatric ECMO circuit. Unlike heparin, 3F7 did not compromise hemostasis. Many ECMO patients are infants who have more unpredictable responses to heparin than do adults or older children. Preventing thrombosis while avoiding bleeding can be difficult to achieve using heparin in this patient population. The study by Larrson et al suggests that interrupting contact activation may be a suitable substitute for heparin in ECMO and perhaps other clinical situations requiring extracorporeal circuits that would not be associated with the hemostatic defect that accompanies heparin therapy. During contact activation, fXIIa converts PK to the kininogenase α-kallikrein, which cleaves HK to liberate the potent vasoactive peptide bradykinin.1 This process likely contributes to inflammation during procedures involving extracorporeal circuits. Inhibition of fXIIa, may have an advantage over inhibition of fXIa in this setting because it would more effectively blunt the contact activation-driven inflammatory responses.
Small-molecule inhibitors targeting fXIa
Several programs are under way to develop small-molecule inhibitors of fXIa that can be administered by parenteral or oral routes. The strategy used in developing the thrombin inhibitors argatroban and dabigatran etexilate and the fXa inhibitors rivaroxaban, apixaban, and edoxaban involves targeting the enzyme active site with a drug.7 The active sites of coagulation factors are located on catalytic domains that are homologs of the digestive enzyme trypsin (Figure 2).5 Conversion of the inactive (zymogen) precursor of a clotting factor to the active protease involves structural changes that result in the enzyme active site adopting an active conformation. Because of the high degree of homology between members of the trypsin family, achieving potent and specific active site inhibition can be difficult. The results of studies with a series of ketoarginine-based peptidomimetics that irreversibly inhibit the fXIa active site illustrate this challenge.27 One compound with an IC50 for fXIa of 6 nM demonstrated an impressive several-hundred-fold better specificity for fXIa than for thrombin or factor Xa. However, the specificity for fXIa was only 2-fold better than for α-kallikrein (a homolog of fXIa) or for trypsin.
BMS-262084 is a 4-carboxy-2-azetidinone-based compound that irreversibly inhibits fXIa by forming a covalent bond with the active site serine residue (Ser557, indicated in yellow in Figure 2).28 This compound produces a dose-dependent antithrombotic effect in rabbit arterial and venous thrombosis models. At a relatively high dose (10 mg/kg/h), the compound increased the cuticle bleeding time by ∼1.5-fold. It is not known if the increased bleeding was specifically due to the loss of fXIa activity or to an off-target effect. Recently, tetrahydroquinalone derivatives that are reversible small-molecule fXIa inhibitors were reported.29 A compound with a Ki of 0.2 nM for fXIa demonstrated >1000-fold selectivity over other plasma proteases, except α-kallikrein (23-fold) and activated protein C (365-fold) and produced a dose-dependent antithrombotic effect in a rabbit arteriovenous shunt thrombosis model without compromising hemostasis.
Hypothetically, for some coagulation proteases, it may be easier to achieve a higher specificity by targeting an area of the enzyme other than the active site. Inhibitors that work through allosteric mechanisms would typically bind to a target enzyme at a site (often referred to as exosites) remote from the active site. Inhibition is produced by induction of conformational changes that compromise protease activity. Investigators at Virginia Commonwealth University reported the characterization of a sulfated pentagalloylglucoside (SPGG) that targets fXIa and allosterically inhibits its activation of factor IX.30 SPGG and other negatively charged sulfated allosteric modulators developed by this group31 are designed to dock initially through the positively charged residues of a polyanion (heparin)-binding site on the fXIa catalytic domain (indicated in blue in Figure 2). Subsequent interactions with other components of the catalytic domain near this binding site are then thought to produce the allosteric inhibitory effect. SPGG appears to be at least 200-fold more selective for fXIa than other clotting factors and effectively prevents factor IX activation by fXIa at submicromolar concentrations. The mechanism of action of this molecule may serve as a model for the development of more potent and specific fXIa inhibitors.
Inhibition of fXIIa with a chimeric protein based on infestin-4
Infestin-4 is a reversible fXIIa active site inhibitor found in the midgut of the blood-sucking insect Triatoma infestan.32 A fusion protein of infestin-4 attached to recombinant human albumin (rHA-Infestin-4) effectively prolonged the aPTT of plasma from mice, rats, rabbits, and humans and is at least 100-fold more selective for fXIIa than for thrombin, kallikrein, and factors VIIa, IXa, Xa, and XIa.32 rHA-Infestin-4 effectively prevented arterial thrombus formation in mice and rats while having no effect on bleeding in a tail bleeding assay. The compound also had an impressive protective effect in mice during cerebral ischemia-reperfusion injury and reduced thrombus size in rat and rabbit arteriovenous shunt thrombosis models. At higher doses of rHA-Infestin-4, the cuticle bleeding time in rats and rabbits was prolonged <2-fold, possibly because of a modest off-target effect on factor Xa.33 Despite this, infestin-4 can serve as a model for the development of even more specific fXIIa inhibitors.
Conclusions and future directions
Observations in the clinic and work with laboratory animals have established a central role for fVIIa/TF-initiated thrombin generation (Figure 1B) in limiting bleeding at a site of blood vessel injury.5,26 In comparison, fXI serves a relatively minor role in this process,2-4 whereas fXII, PK, and HK are not required at all.1,2 The counterintuitive discoveries that mice deficient in a contact protease and baboons treated with anti-fXI antibodies are as resistant (or more resistant) to experimentally induced thrombosis as normal animals treated with full-dose heparin has led us to reevaluate our understanding of the pathogenesis of thrombotic disease. Specifically, the findings present a challenge to the notion that thrombosis simply represents hemostasis in the wrong place and suggest that it may be possible to dissociate antithrombotic and anticoagulant effects to produce drugs that can treat or prevent thromboembolism without placing a patient at risk of severe bleeding. Although the efficacies of fXI and fXII inhibitors have yet to be compared with current therapies, available data suggest scenarios in which such inhibitors may be useful. Data from human populations suggest that targeting fXI or fXIa could have a role in primary or secondary prevention of stroke9 or venous thrombosis.10 Inhibiting fXIa may be particularly useful in patients with high-risk conditions such atrial fibrillation, who are not good candidates for warfarin therapy due to comorbidities such as recent intracranial bleeding. Inhibition of fXIa may also be useful for short-term prophylaxis after neurosurgery and other procedures in which anticoagulant-induced bleeding carries an unacceptable risk. Data from a currently running phase 2 trial testing anti-fXI ASOs as prophylaxis to prevent venous thromboembolism with knee replacement surgery will give us our first indications of the efficacy and safety of targeting fXI as an antithrombotic strategy in humans.
Given the propensity for the contact system to activate on foreign surfaces, treatments targeting fXIa or fXIIa may be particularly useful in situations in which blood comes into contact with artificial components of extracorporeal circuits or indwelling devices. There is a need for alternatives to warfarin for patients with mechanical heart valves. The new oral thrombin and factor Xa inhibitors are not approved for use in these patients, and a phase 2 study (RE-ALIGN)34 comparing dabigatran with warfarin after mechanical heart valve placement was terminated early because of significantly greater incidences of thrombotic and bleeding episodes in the dabigatran arm. The reasons behind the suboptimal performance of dabigatran in this trial have not been established, but variations in plasma drug levels (and particularly subtherapeutic trough levels) and inflammation in the postoperative period have been implicated in contributing to a prothrombotic state.35 An advantage of a therapy targeting fXIa or fXIIa is that it would have a small (or no) impact on bleeding risk. Therefore, the problem of a supratherapeutic plasma level of drug would not be a consideration if the drug is suitably specific for its target. Compounds with long half-lives could be used to safely maintain near complete inhibition of the target protease with little variation across time. Furthermore, in clinical settings such as cardiac surgery, contact activation induced by the extracorporeal oxygenator could be blunted with a fXIIa inhibitor (and perhaps a fXIa inhibitor), reducing the perioperative inflammatory response and its contribution to postoperative thrombotic risk.
It seems clear that increased bleeding places limits on the strategy of combining currently available anticoagulants and/or antiplatelet agents to enhance an antithrombotic effect. In the ATLAS ACS–TIMI 46 trial,36 rivaroxaban was compared with placebo in patients on standard therapy (usually aspirin and a thienopyridine) with recent acute coronary syndrome. Superimposing rivaroxaban on standard therapy significantly reduced death from cardiovascular disease, myocardial infarction, and stroke compared with standard therapy alone, but significantly increased the rate of major bleeding. A therapeutic benefit coupled with increased bleeding was also observed in the TRACER37 and TRA 2P-TIMI 50 trials,38 in which the protease-activated receptor 1 (PAR-1) inhibitor vorapaxar was added to standard therapy (aspirin and a thienopyridine) in patients with acute coronary syndrome or as secondary prevention in patients with atherosclerotic disease, respectively. It is tempting to speculate that adding an inhibitor of fXIa or fXIIa to standard antiplatelet therapy might enhance antithrombotic efficacy in a manner similar to rivaroxaban or vorapaxar, but without the increased incidence of bleeding. It is also possible that the beneficial effects of such inhibitors could be used to reduce the doses of conventional anticoagulants or antiplatelet agents, lowering the baseline bleeding risk associated with the use of these drugs.
Much work remains to be done to establish the types of thromboembolic disorders in humans that involve the contact factors and to establish the effectiveness of strategies targeting these proteases relative to conventional anticoagulation therapy. If effective, however, such drugs could increase the number of patients who are eligible for antithrombotic therapy and widen the spectrum of clinical conditions in which antithrombotic therapy can be safely administered.
Acknowledgments
The author wishes to acknowledge the support of the National Heart, Lung and Blood Institute and the American Heart Association.
Disclosures
Conflict-of-interest disclosure: The author is on advisory committees for Aronora and Isis; has received research funding from Instrument Laboratory; has consulted for Aronora, Bayer, Bristol-Myers Squibb, Dyax, Isis, Merck, and Novartis; and holds patents with or receives royalties from Bayer. Off-label drug use: None disclosed.
Correspondence
David Gailani, MD, Hematology/Oncology Division, Vanderbilt University, 777 Preston Research Building, 2220 Pierce Ave., Nashville, TN 37232; Phone: (615)936-1505; Fax: (615)936-3853; e-mail: dave.gailani@vanderbilt.edu.