Abstract
The myelodysplastic syndromes (MDS) are the most commonly diagnosed myeloid malignancy, with >15 000 new cases identified in the United States yearly. Prognostic scoring systems supplant a formal staging approach and, in general, divide patients into those with lower-risk and those with higher-risk MDS. Although treatment goals for patients with lower-risk disease focus on minimizing transfusions and optimizing quality of life, in higher-risk MDS, the goal is to delay transformation to acute leukemia and to prolong survival. In lower-risk patients, isolated cytopenias are treated with erythropoiesis-stimulating agents or growth factors such as thrombopoietin mimetics. For patients with the del(5q) cytogenetic abnormality or those who fail these initial approaches, lenalidomide may be tried, as can experimental agents. Lower-risk patients with multiple cytopenias may be treated with immunosuppressive drugs or low-dose hypomethylating agents. For patients with higher-risk disease, hypomethylating agents are the preferred initial treatment approach, with evaluation for hematopoietic cell transplantation at diagnosis. Several novel agents are being developed for MDS patients who have failed hypomethylating drugs.
Learning Objective
To describe appropriate therapies for lower- and higher-risk myelodysplastic syndromes and novel agents being developed
Introduction
The myelodysplastic syndromes (MDS) are a heterogeneous collection of clonally derived BM disorders that can cause profound cytopenias and significant compromise in quality and quantity of life. They are the most commonly diagnosed myeloid neoplasms in the United States, with an incidence rate of 4.6/100 000 U.S. citizens, or >15 000 new diagnoses yearly.1 This figure derives from the National Cancer Institute's Surveillance, Epidemiology, and End Results program and the North American Association of Central Cancer Registries and is considered a substantial underestimate, because it is likely compromised by misclassification (50% of patients in such registries, a proportion much higher than the expected rate, are identified as “MDS–unclassifiable”) and underreporting (because the assumption often is made that cytopenias in older adults are a natural, nonmalignant consequence of aging).2
Treatment decisions in MDS depend largely on pathology or prognostic scoring systems appropriated as default staging systems.3 These prognostic systems, the most widely used being the International Prognostic Scoring System (IPSS), are based most commonly on blast percentage, cytogenetic risk groups, and cytopenias, and may also include age, performance status, transfusion needs, and other clinical (and increasingly molecular) factors.4,5 A simpler approach is to divide patients into those with lower-risk or higher-risk disease. Patients with lower-risk MDS fall into IPSS categories of low and intermediate-1, corresponding largely to revised IPSS (IPSS-R) groups very low, low, and sometimes intermediate, and frequently aligning to World Health Organization (WHO) histologic subtypes refractory cytopenia with unilineage or multilineage dysplasia or with ring sideroblasts, MDS with del(5q), or MDS unclassifiable. For these patients, median overall survival can be measured in years. Patients with higher-risk disease fall into IPSS categories of intermediate-2 and high groups, corresponding largely to IPSS-R groups very high, high, and sometimes intermediate, or to WHO histologic subtypes of refractory anemia with excess blasts (RAEB)-1 and RAEB-2, with an expected median overall survival of <2 years.3,6,7 Highlighting the severity of these diseases, if we accept the premise that the IPSS is a default MDS staging system, with low-high reflecting stages 1-4, and comparing it stage-for-stage with American Joint Committee on Cancer staging for non-small-cell lung cancer, overall survival is worse for MDS.8,9
Therefore, the first step in initiating a discussion with a patient about treatment is to make sure that the patient understands the severity of the disease (because this is commonly downplayed at diagnosis) and that patient and physician goals are aligned.10,11 As a general statement, treatment goals for patients with lower-risk disease focus on minimizing transfusions and optimizing quality of life. Because no therapy for lower-risk disease has been shown prospectively to improve overall survival, in asymptomatic patients with relatively preserved blood counts, treatment initiation should be delayed as long as possible. In contrast, for higher-risk patients, in whom survival is severely truncated and transformation to acute myeloid leukemia (AML) is likely and akin to a death knell,3,6 treatment should be started at diagnosis regardless of peripheral counts.
Therapies for lower-risk MDS
Initial treatment of isolated cytopenias
Most lower-risk MDS patients initiate their diagnostic evaluation as a result of cytopenias detected on routine blood work, the most common of which is anemia. Therefore, therapies focus on alleviating those cytopenias (Figure 1). This is initially accomplished most easily through transfusions (for anemia or thrombocytopenia) or through the use of erythropoiesis-stimulating agents (ESAs) or growth factors.

Treatment algorithm for lower-risk MDS. *Thrombopoietin agonists should only be used outside of a clinical trial in the setting of immune-mediated thrombocytopenia
Treatment algorithm for lower-risk MDS. *Thrombopoietin agonists should only be used outside of a clinical trial in the setting of immune-mediated thrombocytopenia
Of all patients with lower-risk disease and anemia, ∼40% will achieve an International Working Group (IWG)-defined hematologic improvement to ESAs for a median duration of 2 years.12,13 A model developed by the Nordic MDS group identifies patients more likely to respond to ESAs. Those with low or absent transfusion requirements (<2 pRBC units/month) and a low serum erythropoietin level (<500 units) have a higher response rate (74%) than those with high transfusion needs (≥2 units/month) and serum erythropoietin levels (≥500 units, or 7%).14 One decision analysis has shown that patients falling into the former group live longer and have improved quality of life if treated initially with ESAs, whereas those in the latter group have improved outcomes if treated initially with non-growth-factor approaches.15 The IPSS-R, too, can predict response to ESAs, with 85% of patients in one study in the very low-risk group responding, compared with 31% of patients in the high-risk group.16
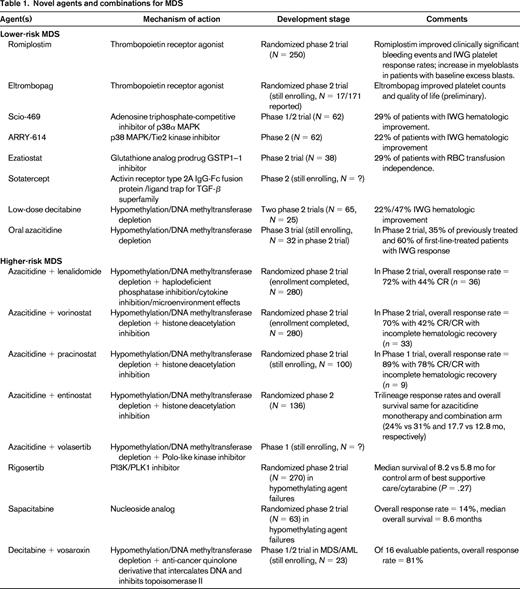
Lower-risk MDS patients with thrombocytopenia have been treated within the context of clinical trials with the thrombopoietin receptor agonists romiplostim and eltrombopag (Table 1). The largest study randomized 250 lower-risk MDS patients with thrombocytopenia in a 2:1 fashion to receive romiplostim or placebo. The number of clinically significant bleeding events favored patients receiving romiplostim, significantly so in those with baseline platelet counts ≥20 × 109/L (relative risk = 0.35, P < .0001), as did IWG platelet response rates (odds ratio = 15.6).17 A model similar to the Nordic MDS model for ESAs has also been developed and validated in romiplostim patients to predict response and reached similar conclusions based on baseline platelet transfusion needs and serum thrombopoietin levels. The study was stopped early due to concern by the data monitoring committee of an increased rate of AML evolution and an increase in myeloblasts seen in the romiplostim group [hazard ratio (HR) = 2.5]. However, with further follow-up, these rates equalized and had no impact on overall survival (HR = 0.86 favoring romiplostim). Given these concerns, the use of either romiplostim or eltrombopag should be avoided in patients with excess blasts.
Presumptive use of granulocyte (or granulocyte macrophage) colony stimulating factors for MDS patients with neutropenia has never been shown prospectively to reduce episodes of febrile neutropenia or to improve survival and is discouraged.
Treatment of anemia in del(5q) MDS and in non-del(5q) MDS after ESA failure
Lenalidomide is approved by the U.S. Food and Drug Administration (FDA) for the treatment of lower-risk, transfusion-dependent MDS patients with the del(5q) cytogenetic abnormality. Its activity is thought to be mediated through selective suppression of del(5q) clones by inhibition of haplodeficient phosphatases encoded within or near the proximal common deleted region that releases progenitors from p53 arrest, followed by terminal arrest and apoptosis at G2/M, in association with a defect in ribosomal protein function.18,19 In non-del(5q) anemia MDS patients, lenalidomide likely modulates the BM microenvironment, inhibits lysine demethylase, and potentiates erythropoietin signaling.
In the phase 2 registration study conducted in del(5q) patients,20 148 were treated with lenalidomide, of whom 99 (67%) achieved transfusion independence, including every patient who experienced a cytogenetic response. With a median follow-up of 3.2 years, median response duration was 2.2 years and median overall survival was 3.3 years, longer in patients with durable transfusion independence responses (4.3 vs 2.0 years in nonresponders; P < .0001) and in cytogenetic responders (4.9 vs 3.1 years in nonresponders; P = .010). A subsequent phase 3, double-blind trial of lenalidomide at 2 different doses versus supportive care in a similar population demonstrated transfusion independence responses lasting >6 months in 43%–52% of subjects, compared with 6% of controls, and a cytogenetic response rate in lenalidomide-treated subjects of 25%–50%.21 Lower IPSS-R category and cytogenetic response to lenalidomide has since been shown to be associated with durable transfusion independence and reduced risk of AML transformation or death, whereas treatment-related thrombocytopenia has been associated with response to lenalidomide.
In anemic, lower-risk MDS patients who do not harbor the del(5q) lesion, a phase 2 study of lenalidomide similar in design to the registration study enrolled 215 patients.22 Approximately one-quarter (26%) achieved RBC transfusion independence. Duration of response was less than what is expected for del(5q) patients, at a median of 41 weeks (range, 8-136). Although grade 3 or 4 myelosuppression was approximately one-half of that seen in the registration study, at 20%–25%, it also was not associated with subsequent attainment of a transfusion independence response to therapy.
Although concern has been raised regarding the risk of secondary malignancies after lenalidomide exposure, an analysis of combined data from the phase 2 and 3 studies in 286 del(5q) patients treated with lenalidomide showed a rate of AML evolution of 17% at 2 years (with a median follow-up of 3 years), whereas another analysis of 381 untreated del(5q) MDS patients, with a median follow-up of ∼4 years, showed a rate of AML evolution of 17% at 5 years.23,24
Several novel agents have been explored in clinical trials in anemic, lower-risk MDS patients. Scio-469, a potent selective adenosine triphosphate-competitive inhibitor of p38α MAPK, was administered to 62 patients in a phase 1/2 study, of whom 18 (29%) experienced an IWG-defined hematologic improvement. ARRY-614, another p38 MAPK/Tie2 kinase inhibitor, was administered to 62 lower-risk MDS patients. Of 54 evaluable patients, 12 (22%) had an IWG-defined hematologic improvement. Ezatiostat, a glutathione analog prodrug glutathione S-transferase P1-1 (GSTP1-1) inhibitor, was explored in the phase 2 setting in 38 lower-risk MDS patients, of whom 11 (29%) achieved RBC transfusion independence lasting a median of 34 weeks.25,26 Sotatercept, a soluble activin receptor type 2A IgG-Fc fusion protein that acts as a ligand trap for members of the TGF-β superfamily, is undergoing multicenter trials in MDS.
Treatment of multiple cytopenias
Immune dysregulation contributes to ineffective hematopoiesis in a subset of MDS patients. Although, classically, these patients have been identified as those with hypocellular BMs, in truth, factors predictive of a higher likelihood of response to immunosuppressive therapy include the HLA-DR15 genotype, younger age, lack of transfusion dependence, the presence of a paroxysmal nocturnal hemoglobinuria clone, and normal karyotype or trisomy 8.
In a multicenter, phase 3 trial, 88 patients (the majority of whom had lower-risk MDS) were randomized to combined antithymocyte globulin + cyclosporine or to best supportive care.27 The complete (CR)/partial (PR) remission response rate in patients receiving immunosuppressive therapy was 29%, with a median response duration of 16.4 months, compared with 10% in the best supportive care arm (P = .02). Survival did not differ between arms. The largest U.S. experience is a phase 2 multicenter study of antithymocyte globulin administered to 27 MDS patients, the majority of whom had lower-risk disease.28 The IWG-defined response rate was 33%, with a median response duration of 245 days. In a multivariate model, shorter disease duration, having a higher proportion of CD8+ terminal memory cells, and a higher CD4+ T-cell proliferative index (Ki67+) independently discriminated response.
So-called hypomethylating agents (azacitidine and decitabine) are also used in lower-risk MDS patients with multiple cytopenias, frequently after failure of other agents. These will be covered in more detail in the section on therapies for higher-risk MDS. In lower-risk patients, IWG response rates appear to be 30%–40%. Low-dose decitabine has been explored in 2 phase 2 studies, the first of which (giving decitabine at a dose of 20 mg/m2 on 3 days per 28-day cycle) enrolled 65 lower-risk MDS patients, of whom 22% achieved an IWG response.29 The second study (which administered decitabine at 3.5-7 mg/m2 1-3 times weekly) enrolled 25 patients, of whom 47% achieved an IWG response.30 Oral azacitidine has been explored in one study of 41 patients, of whom 48% had lower-risk disease. Among the 32 enrolled MDS patients, 35% of previously treated and 60% of first-line-treated patients had an IWG response excluding BM responses. This drug is being explored in the phase 3 setting.31 Rigosertib, a dual PI3 and polo-like kinase inhibitor, is also being studied as an oral formulation in lower-risk MDS.
Therapies for higher-risk MDS
Initial treatment of higher-risk MDS
Unlike the case with lower-risk MDS, the prospect of reduced survival and brisk disease progression warrant the initiation of disease-modifying therapy at diagnosis with higher-risk MDS (Figure 2). In common practice, this often takes the form of one of the hypomethylating agents, azacitidine or decitabine. The postulated mechanism of action is rooted in reversing the aberrant methylation of tumor suppressor genes common in higher-risk MDS via protesomic destruction by way of depleting DNA methlytransferase.32,33 Unearthing of recurrent gene mutations in DNMT3A, TET2, IDH1 and IDH2, ASXL1, and others critical in DNA methylation and posttranslational modification of histones reinforces the essential role of epigenetic regulation in MDS leukemogenesis and establishes the basis for using an epigenetic strategy in patients harboring these abnormalities.34 Despite this understanding, alterations in DNA methylation patterns have not been consistently linked to outcomes and a validated, prospective biomarker that can reliably predict response remains elusive.

Treatment algorithm for higher-risk MDS. Reprinted with permission from Sekeres and Cutler, 2014.47
Treatment algorithm for higher-risk MDS. Reprinted with permission from Sekeres and Cutler, 2014.47
Azacitidine gained approval from the FDA based on a phase 3 trial in MDS patients of all subtypes (including higher-risk MDS patients and lower-risk patients with significant cytopenias) in which it was compared with supportive care.35 In this study, which allowed for crossover, azacitidine was associated with a 14% CR and PR, as well as a 30% hematologic improvement when evaluated using IWG criteria, but not with an improved overall survival. In the subsequent AZA-001 study, which was limited to higher-risk MDS patients, when azacitidine was pitted against a predetermined conventional care regiment (supportive care, low-dose cytarabine, or traditional, AML-like induction chemotherapy), it did result in an overall survival advantage, with a median survival of 24.5 versus 15 months for conventional care regimens after a median follow-up of 21.1 months (HR = 0.58, P = .0001).36
Likewise, decitabine gained regulatory approval in the United States based on a phase 3 trial in which it was compared with supportive care.37 By IWG criteria, 17% of patients attained remission (CR and PR) and 13% were found to have hematologic improvement. Despite showing response rates similar to azacitidine, a survival advantage over best supportive care was not demonstrated in a randomized controlled trial in higher-risk MDS patients, with a median overall survival for decitabine-treated patients of 10.1 months compared with 8.5 months for best supportive care patients (HR = .88, P = .38).38
Why was a survival difference demonstrated for 1 hypomethylating agent but not the other when the 2 are structurally and biologically so similar? Given the vast differences in median survival on the control arms, which cannot be explained by the 30% of patients in the azacitidine study on the conventional care arm who received active treatment, it is highly likely that different patient populations were enrolled in each study. In addition, the median number of cycles of drug received was 9 for azacitidine and 4 for decitabine, which may have been inadequate to realize all responses; in addition, decitabine may have been administered using a suboptimal schedule. No study has randomized patients to hypomethylating therapy cessation soon after maximal response versus treatment ad infinitum for as long as it is working. Any attempts to study this retrospectively would likely be corrupted by confounding and no prospective study has reported “recovering” responses to adequate hypomethylating therapy upon relapse with resumption of the same or a different hypomethylating drug (eg, switching from azacitidine to decitabine). Therefore, the standard is to continue a hypomethylating agent for as long as a response persists and to avoid switching drugs.
Combination therapies
Building on the single-agent success of hypomethylating agents, combination therapy is a logical next step, with the rationale based primarily on tumor cell heterogeneity and its implication for drug resistance, in vitro synergy of agents, and the success of combination chemotherapy in other hematologic malignancies such as acute leukemias and lymphomas. Azacitidine and lenalidomide have complementary mechanisms of eliciting a response in patients with MDS. The observation that patients with higher-risk MDS retain features similar to lower-risk disease—both microenvironment and cell-regulatory mechanisms likely play a role in progression—provided the impetus for combining these 2 agents. The combination was tested in a multicenter phase 1/2 study of 36 patients treated with the combination, of whom 16 (44%) achieved a CR and 10 (28%) experienced hematologic improvement for an overall response rate of 72%.39 At a median follow-up of 12 months, the overall survival was 13.6 months for the entire cohort and >37 months for the subgroup that achieved a CR.
Although histone deacetylase (HDAC) inhibitors have led to modest results in the single-agent setting, the prospect of an additive or synergistic effect with hypomethylating therapy has driven a series of current trials. Vorinostat, a small-molecule inhibitor of both class I and II HDAC enzymes, promotes cell-cycle arrest and apoptosis of cancer cells through regulation of gene expression. The outcomes of 40 primarily higher-risk patients enrolled in a phase 2 trial testing the combination of vorinostat and azacitidine highlight the potential of combined epigenetic therapy.40 Of the 33 patients evaluable, 23 (70%) responded, with a CR (CR/CR with incomplete hematologic recovery) in 14 and an additional 9 with hematologic improvement. The median duration of response was 16 months and the median overall survival was 21 months. Despite a large majority of patients with undetectable disease by morphology, analysis for the MDS clone by FISH and cytogenetics at the time of best response revealed a persistent clone in 45% of cases, suggesting a mechanism of action via disease modulation rather than direct cytotoxic clearing of the malignant clone. The North American Intergroup MDS study (S1117, www.clinicaltrials.gov identifier NCT01522976) is testing this combination, along with the combination of lenalidomide and azacitidine, against single-agent azacitidine in the front-line, higher-risk setting.
Pracinostat is an oral pan-HDAC inhibitor that has also demonstrated synergistic interactions with azacitidine in preclinical studies. After a successful single-agent study in myelofibrosis, a pilot phase 2 study of pracinostat combined with azacitidine has been conducted in 9 patients with advanced MDS, in whom the overall response rate was 89% (7 achieved CR/CR with incomplete hematologic recovery).41 Furthermore, 5 patients (56%) achieved a complete cytogenetic response. Invigorated by these results, a randomized placebo-controlled phase 2 trial of the combination (www.clinicaltrials.gov identifier NCT01873703) has been launched through the MDS Clinical Research Consortium.
A cautionary tale in combination therapy is seen in the phase 2 exploration of the HDAC inhibitor entinostat combined with azacitidine versus azacitidine monotherapy, which enrolled 136 higher-risk MDS and AML patients.42 Unfortunately, rates of trilineage response, the primary outcome, and median overall survival were similar for those treated with monotherapy (on a 10-day schedule) and those treated with azacitidine combined with the HDAC inhibitor entinostat (24% vs 31% and 17.7 vs 12.8 months, P = .15, respectively), although response rates were higher for the 10-day azacitidine schedule than for historic controls treated on a 7-day schedule.
Allogeneic hematopoietic stem cell transplantation
One fact distinguishing AML from MDS is that neither single-agent nor combination therapies can cure MDS regardless of disease risk, whereas AML can be cured with cytotoxic chemotherapy. The only treatment modality that has resulted in long-term, maintenance-free remission is allogeneic hematopoietic stem cell transplantation (HSCT). Although HSCT for MDS is reviewed in detail elsewhere in this publication, it is critical to mention here because HSCT and non-HSCT therapies are often integrated in a given patient's care. Most patients undergo some form of pre-HSCT therapy to delay progression to leukemia and to reduce disease burden. Although several retrospective studies have explored various pre-HSCT therapies,43,44 the optimal approach has yet to be defined. A prospective trial randomizing HSCT-eligible patients to hypomethylating therapy versus induction chemotherapy before HSCT is under way (www.clinicaltrials.gov identifier NCT01812252). Furthermore, several preemptive and prophylactic strategies are being explored to improve post-HSCT outcomes by reducing relapse. Because peri-HSCT therapies are optimized with conditioning regimens, complete MDS treatment packages will likely emerge (Figure 3) and, because pre-HSCT and post-HSCT therapies must be integrated with the HSCT regimen, early consultation and collaboration with a transplantation specialist should be routine.

Peritransplantation management of MDS. Reprinted with permission from Gerds and Scott, 2012.48
Peritransplantation management of MDS. Reprinted with permission from Gerds and Scott, 2012.48
Treatment after hypomethylating agent failure
Only 3 drugs have received regulatory approval specifically for the treatment of MDS, all with suboptimal response rates at <50% of limited durability, typically 1-2 years. Once these agents are no longer effective, there is no standard of care established for second-line treatment (eg, the relapsed/refractory setting). Furthermore, prognosis after hypomethylating agent failure is dismal, with median survival estimated at <6 months for higher-risk patients, and <18 months for lower-risk patients.45 Several resistance mechanisms are being targeted in an attempt to overcome the limitations of current therapies.
One such approach takes advantage of the relationship between immune dysregulation and the development of MDS, which may play a role in resistance to standard therapies. Select MDS patients can experience a CR to immunosuppressive therapy, and self-tumor antigens on the surface of the MDS cells within the niche may also induce the preferential proliferation of CD4+CD25+FOXP3+ T-regulatory cells. Recently, aberrant up-regulation of PD-1, PD-1L, and CTLA4 in MDS has been described, specifically PD-L1 protein expression in CD34+ MDS cells, whereas the stroma/nonblast cellular compartment was positive for PD-1, the signaling of which may be specific to hypomethylating agent resistance.46
The largest trial for this indication randomized patients in a 1:2 fashion (N = 270) to receive placebo or the PI3K/PLK1 inhibitor rigosertib. There was no significant overall survival benefit in patients receiving rigosertib, with a median survival of 8.2 months compared with 5.8 months for the control arm (P = .27). Other agents being explored in this context include the nucleoside analog sapacitabine, which had an overall response rate of 14% and median overall survival of 8.6 months in MDS patients failing hypomethylating agents; the polo-like kinase inhibitor volasertib; and the quinolone derivative vosaroxin.
Summary and conclusions
MDS treatment is risk stratified, with risk defined by cytopenias, blast percentage, and traditional metaphase cytogenetics. Because, on average, 9 somatic mutational events occur that lead to the MDS clinical phenotype, it is no wonder that a “magic bullet” drug therapy has not yet been discovered. Nevertheless, several agents directed at improving cytopenias without the cost of side effects are being developed in lower-risk MDS and have the potential to improve quality of life for patients with more indolent disease. Combination therapies, often including an approved plus a novel agent, are at the forefront of treatment for higher-risk disease and are poised to make an immediate impact on the treatment landscape. Without doubt, the next regulatory frontier lies in treatment of patients who have failed hypomethlylation agent therapy. A molecular revolution is taking place that will undoubtedly redefine MDS biology, classification, risk, and therapy in the coming decade.
Disclosures
Conflict-of-interest disclosures: M.A.S. is on the board of directors or an advisory committee for Celgene, Amgen, and Boehringer Ingelheim. A.T.G. declares no competing interests. Off-label drug use: Lenalidomide for non-del(5q) and higher-risk MDS; vorinostat for higher-risk MDS.
Correspondence
Mikkael A. Sekeres, MD, MS, Leukemia Program Cleveland Clinic Taussig Cancer Institute, Desk R35, 9500 Euclid Avenue, Cleveland, OH 44195. Phone: 216-445-9353; Fax: 216-636-0636; e-mail: sekerem@ccf.org.