Abstract
Now that the spectrum of somatic mutations that initiate, propagate, and drive the progression of myeloproliferative neoplasms (MPNs) has largely been defined, recent efforts have focused on integrating this information into clinical decision making. In this regard, the greatest progress has been made in myelofibrosis, in which high-molecular-risk mutations have been identified and incorporated into prognostic models to help guide treatment decisions. In this chapter, we focus on advances in 4 main areas: (1) What are the MPN phenotypic driver mutations? (2) What constitutes high molecular risk in MPN (focusing on ASXL1)? (3) How do we risk-stratify patients with MPN? And (4) What is the significance of molecular genetics for MPN treatment? Although substantial progress has been made, we still have an incomplete understanding of the molecular basis for phenotypic diversity in MPN, and few rationally designed therapeutic approaches to target high-risk mutations are available. Ongoing research efforts in these areas are critical to understanding the biological consequences of genetic heterogeneity in MPN and to improving outcomes for patients.
Learning Objectives
Understand the role played by myeloproliferative neoplasm (MPN) phenotypic driver mutations and concomitant somatic mutations in the development of MPN
Discuss phenotypic diversity in MPN and understand the effect of molecular analysis on the biology, prognosis, and treatment of MPN
Myeloproliferative neoplasm case
A 61-year-old man presents with mild fatigue and is noted to have an abnormal complete blood count demonstrating leukocytosis (white blood cells, 15 ×109/L [neutrophils, 65%; lymphocytes, 20%; monocytes, 15%; eosinophils, 0%; basophils, 0%), mild anemia (hemoglobin, 10.5 g/dL; mean corpuscular volume, 90 fL), and thrombocytosis (platelets, 600 ×109/L). Comorbidities include hypertension and hyperlipidemia. On physical examination, the spleen is palpable 4 cm below the left costal margin. The peripheral blood smear reveals rare dacrocytes and leukoerythroblastosis. There are 0% peripheral blood blasts. A bone aspirate and marrow biopsy is consistent with a diagnosis of primary myelofibrosis (PMF) and indicates myelofibrosis-2 (MF-2) grade fibrosis. The karyotype is normal. A next-generation sequencing (NGS) panel reveals the presence of a CALR type 1 mutation (variant allele fraction = 45%) and an ASXL1 Y588X mutation (variant allele fraction = 35%). No other mutations are detected.
Introduction
In recent years, with the expanded use of NGS technologies, we have come to fully appreciate the molecular complexity of myeloproliferative neoplasms (MPNs), a group of clonal diseases arising in the hematopoietic stem cell (HSC) compartment, resulting in the overproduction of terminally differentiated cells of the myeloid lineage. The integration of molecular genetics into prognostication models and treatment decisions is now firmly established in myelofibrosis (MF) and a similar paradigm is likely to follow in the coming years in polycythemia vera (PV) and essential thrombocythemia (ET), although large prospective studies with long follow-up will be required to fully decipher the effect of genetic heterogeneity in these diseases. In this review, we focus on 4 questions surrounding the molecular genetics of MPN. (1) What are the MPN phenotypic driver mutations? (2) What constitutes high molecular risk (HMR) in MPN (focusing on ASXL1)? (3) How do we risk-stratify patients with MPN? (4) What is the significance of molecular genetics for MPN treatment?
What are the MPN phenotypic driver mutations?
In >95% of cases of MPN, the mutations that drive the development of an MPN phenotype are accounted for by somatic mutations in 3 genes: JAK2, CALR, or MPL; notably, these mutations occur in a mutually exclusive manner.1 Mutations in JAK2 and MPL occur as gain-of-function point mutations (ie, JAK2 V617F and MPL W515L/K, respectively), whereas the mutations in CALR occur as +1 base pair (bp) frameshifts in the last coding exon of CALR, which result in the generation of a novel C terminus.1,2 A number of lines of evidence support the proposition that mutations in JAK2, CALR, or MPL initiate MPN and are sufficient alone to engender a full MPN disease phenotype. Sequencing studies of MPN patients provide the first evidence. In ∼50% cases of MPN, a mutation in JAK2, CALR, or MPL is the sole mutation identified based on our current level of knowledge of genes known to be somatically mutated in myeloid malignances.3 Furthermore, JAK2 V617F is one of the most common mutations associated with the development of clonal hematopoiesis of indeterminate potential (CHIP), an entity in which clonally restricted somatic mutations in genes associated with hematological malignancies are found in normal individuals.4 In virtually all cases of JAK2 V617F+ CHIP, the JAK2 mutation occurs as the sole genetic event, suggesting that JAK2 V617F is an early disease-initiating event and indicating that JAK2 V617F alone is sufficient to engender clonal hematopoiesis.5 The prevalence of JAK2 V617F+ MPN is significantly lower than that of JAK2 V617F+ CHIP, suggesting that JAK2 V617F+ CHIP does not always progress to MPN. Recent publications indicate that JAK2 V617F+ CHIP is itself a clinically relevant entity, being associated with an increased risk of both coronary heart disease6 and venous thrombosis.7 Further evidence that mutations in JAK2, CALR, or MPL are sufficient to engender an MPN phenotype has been provided by mouse models, where expression of each mutation alone is sufficient to induce MPN.8-10
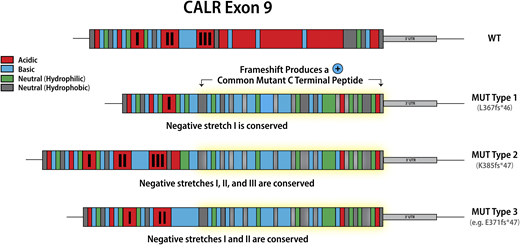
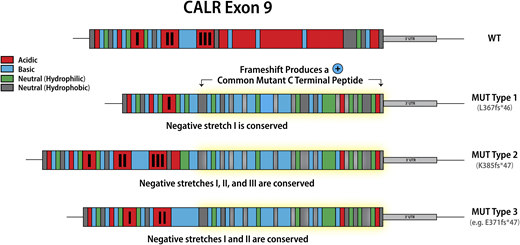
Classification of calreticulin mutations in MPN
Mutations in calreticulin (CALR), an endoplasmic reticulum chaperone, occur as heterozygous insertions and/or deletions in exon 9 of the gene. Although >50 different CALR mutations have been identified in MPN patients, all result in a +1-bp reading frameshift and the generation of a mutant-specific 36 amino acid C-terminal tail. Mutant CALR has been demonstrated to bind the thrombopoietin receptor MPL to activate JAK-STAT signaling in a thrombopoietin-independent manner, with the positive charge of the C terminus of the mutant CALR protein and its lectin-binding residues required for its oncogenic activity.11,12 CALR mutations were originally classified as type 1 (52-bp deletion) and type 2 (5-bp insertion) on the basis that these mutations are the most common, accounting for ∼50% and 30% CALR mutations, respectively2 . This classification was later refined to encompass (1) type 1 and type 1–like (65%), (2) type 2 and type 2–like (32%), and (3) other (3%) groups, with these categories defined on the basis of the deletion of stretches of negatively charged amino acids in the wild-type CALR C terminus.13 Type 1 and type 1–like mutations result in the deletion of 2 stretches of negatively charged amino acids; type 2 and type 2–like mutations do not result in the deletion of negatively charged amino acids, and other mutations result in the deletion of 1 stretch of negatively charged amino acids (Figure 1). The classification of CALR mutations is relevant not just in terms of the change in the composition of the CALR C terminus, but also has implications for both MPN phenotype and prognosis, suggesting that despite all CALR mutations resulting in loss of the KDEL sequence and the generation of a common mutant-specific C-terminal peptide tail, they are not biologically equivalent. Type 1 and type 1–like CALR mutations are more common in MF than in ET13,14 and, when present in ET, are associated with an increased risk of myelofibrotic transformation.13 Type 2 and type 2–like CALR are more common in ET than in MF, but when present in MF are associated with a worse prognosis compared with type 1 and type 1–like mutations.15
What constitutes HMR in MPN?
In addition to the MPN phenotypic driver mutations, patients with MPN frequently harbor concomitant mutations in other genes commonly mutated across myeloid malignancies more broadly. These include mutations in genes involved in epigenetic regulation (eg, TET2, ASXL1, DNMT3A, EZH2, IDH1/2) and RNA splicing (SRSF2, SF3B1, U2AF1), in addition to mutations in cancer-associated genes (eg, p53). With the increasing use of NGS panels clinically, concomitant nonphenotypic driver mutations are gaining relevance in terms of prognostic significance. Among the nonphenotypic driver mutations, ASXL1 in particular, but also EZH2, IDH1/2, and SRSF2 have a strong impact on prognosis in MF,16 in which the presence of mutations in any of these genes defines HMR.17 The number of mutations also holds prognostic significance in MF, with multiple mutations having a worse prognosis independent of the specific mutation.16
Role of ASXL1 mutations in MPN
Additional sex combs–like transcriptional regulator 1 (ASXL1) is one of the most frequently mutated genes in MPN.1 Depending on the cohort analyzed, it can be detected in up to 35% of patients with PMF.18 Mutations in ASXL1 localize to 5′ end of the last exon (exon 12) are mainly frameshift and occasionally nonsense,18 resulting in the expression of a C-terminal truncated ASXL1 protein. The role of mutations in ASXL1 has been debated but recent data using a transgenic mouse model suggest that ASXL1 mutations are gain-of-function,19 a finding that is supported by ASXL1 mutations tending to be heterozygous with the wild-type allele retained.
Mechanism by which ASXL1 mutations cause myeloid malignancies
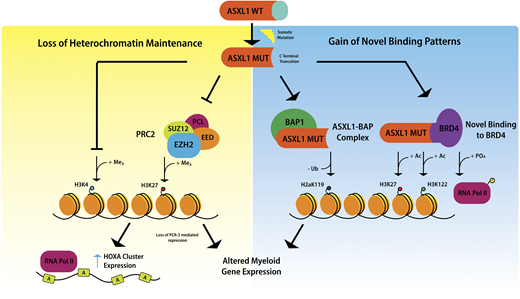
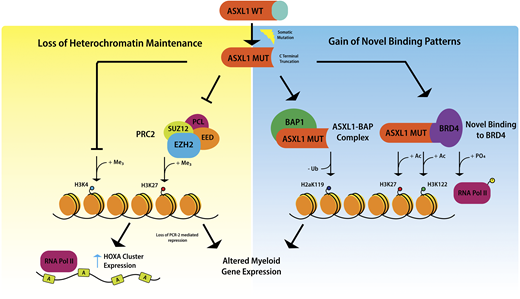
Mechanistic studies of ASXL1 in myeloid malignancies have shown that mutations in ASXL1 result in loss of polycomb repressive complex 2–mediated histone H3 lysine 27 (H3K27) tri-methylation, and the consequent loss of repression of the HOXA cluster.20 Recent data indicate that mutations in ASXL1 may also result in inhibition of H3K4 methylation.21 It has been suggested that ASXL1 truncation mutations confer gain-of-function through enhancing the activity of the ASXL1-BAP deubiquitinase complex, resulting in loss of histone H2AK119 ubiquitination, which together with loss of H3K27 tri-methylation, upregulates genes involved in myeloid differentiation.22 Recently, mutant ASXL1 was shown to directly bind BET bromodomain-containing protein 4 , causing phosphorylation of RNA polymerase II and acetylation of H3K27 and H3K122, again leading to upregulation of genes with a role in myeloid differentiation19 (Figure 2).
Mouse models of mutant ASXL1
Initially, ASXL1 mutations were modeled in mice through the generation of a conditional knockout. Hematopoietic-specific loss of Asxl1 resulted in impaired HSC self-renewal capability, pancytopenia, and myelodysplastic syndrome (MDS).23 These initial data supported the idea that ASXL1 was a loss-of-function mutation because it was required for normal hematopoiesis and its loss could confer a myeloid malignancy phenotype.
Recently, constitutive knock-in mice of a common ASXL1 frameshift mutation (G643WfsX12) have been generated.24,25 In 1 model, Asxl1G643fs/+ mice developed a hematopoietic phenotype consistent with MDS and died at ∼600 days from a disease reminiscent of human chronic myelomonocytic leukemia.24 Mechanistic studies in this model revealed derepression of genes normally repressed by BMI1, including p16Ink4a.24 Heterozygous Asxl1G643fs/+ mice showed decreased body size and weight similar to the phenotype described in patients affected by Bohring-Opitz syndrome, which is caused by a germline ASXL1 mutation. In the other model, heterozygous Asxl1G643fs/+–expressing mice did not acquire any defined myeloid malignancy and had reduced repopulation capacity in vivo but enhanced serial replating activity in vitro.25 The reason for the difference in phenotype for these 2 models is not readily apparent. More recently, a transgenic mouse model expressing a truncated ASXL1 protein from the Vav promoter (Asxl1Y588X Tg) was described.19 These mice acquired a large spectrum of myeloid malignancies including MPN, MDS, acute myeloid leukemia, and MDS-MPN. mainly after 8 months of age.19 The mice with MPN phenotypes (36.4% of the cohort) showed higher platelets counts, enlarged spleens, extramedullary hematopoiesis (in spleen and liver). and megakaryocytic hyperplasia.19 Asxl1Y588X Tg mice displayed an expanded HSC compartment and enhanced HSC self-renewal, whereas bone marrow cells derived from these mice showed an increased susceptibility to BET bromodomain inhibition.19 Another transgenic mouse model (Rosa26 locus), which models the human ASXL1 frameshift mutation (E635RfsX15), and again results in expression of a C-terminal truncated ASXL1 protein, was also recently described.26 These mice developed myeloid skewing, age-dependent anemia, thrombocytosis, and evidence of morphological dysplasia, whereas HSC from these animals showed a competitive disadvantage. Reductions in H3K4me3 and H2AK119Ub without significant reductions in H3K27me3 were seen.
What is the clinical significance of an ASXL1 mutation?
Mutations in ASXL1 have been linked to adverse prognosis in PMF,17,27 where they are associated with older age and increased white blood cell count.18 ASXL1 mutations do not segregate with a particular MPN phenotypic driver mutation, and in CALR-mutant MF it has been observed that ASXL1 mutations are more associated with a CALR type-1 mutation than with a CALR type-2.18 The favorable prognosis of CALR-mutant PMF has been shown to be negatively affected by the presence of an ASXL1 mutation.28 In MF, patients harboring an ASXL1 mutation had a shorter time to ruxolitinib treatment failure and reduced overall survival.29 It has also been reported that ASXL1 is the gene that most frequently acquired mutations in MF during ruxolitinib treatment, and the acquisition of an ASXL1 mutation was associated with the development of leukocytosis and thrombocytopenia.30 Whether MPN subclones harboring an ASXL1 mutation are positively selected for during ruxolitinib treatment remains to be determined. Finally, ASXL1 mutations were also demonstrated to have an adverse prognosis in MF in the context of allogeneic stem cell transplantation, where they were an independent risk factor for lower progression-free survival.31
How do we risk-stratify MPN patients?
Risk stratification in ET and PV
In ET and PV, risk stratification has been developed primarily to estimate the thrombotic risk. In PV, age and prior history of thrombosis remain the factors that define thrombotic risk using the conventional low- and high-risk stratifications.32 In ET, molecular genetics has been integrated into thrombotic risk stratification. The International Prognostic Score of Thrombosis for Essential Thrombocythemia (IPSET)-thrombosis (International Prognostic Score in ET) model added the presence of the JAK2 V617F mutation and cardiovascular risk factors to conventional risk stratification to define 3 risk groups: low, intermediate, and high.33 Recently, IPSET-thrombosis was further revised with the addition of a very-low-risk group possessing no adverse risk factors for thrombosis.34 The revised IPSET-thrombosis model now defines 4 risk categories based on 3 adverse variables (thrombosis history, age >60 years, and JAK2 V617F): very low (no adverse features), low (presence of JAK2 V617F), intermediate (age >60 years), and high (presence of thrombosis history or presence of both advanced age and JAK2 V617F).34 Currently, aspirin (81-100 mg daily) is recommended in intermediate- or high-risk groups, and aspirin (81-100 mg daily) or observation is recommended in the very low or low-risk groups.35 Recent data suggest that the specific MPN phenotypic driver mutation should be considered in terms of deciding on the use of aspirin, with 1 recent retrospective study indicating that anti-platelet therapy in CALR-mutant low-risk ET did not reduce thrombosis and was associated with an increased risk of bleeding.36 Currently, cytoreductive therapy is recommended in the high-risk group.35 Prospective clinical trials are required to more definitively determine the best risk-adapted therapy in each of the revised IPSET-thrombosis groups.
NGS studies to define molecular risk in ET and PV are starting to emerge.37 In 1 recent study, in PV an ASXL1 mutation was associated with a lower OS and an SRSF2 mutation with a lower MF-free, leukemia-free, and OS.37 In ET, IDH2 and SH2B3 mutations were associated with lower OS, TP53 mutations with lower leukemia-free survival, and SF3B1 and U2AF1 mutations with higher MF transformation.37 These findings will need to be validated in larger independent cohorts, and ultimately prospective studies will be required to fully determine the effect of molecular genetic abnormalities in PV and ET.
Effect of molecular genetics on risk stratification in MF
The World Health Organization created a new diagnostic category in 2016 that split patients with MF into 2 separate groups: prefibrotic-MF and overt-MF.38 Some patients previously classified as having ET now fall into the prefibrotic-MF group, and it has been demonstrated that prefibrotic-MF has a different prognostic outcome from both ET and MF.39
Over the past 10 years, prognostic scoring systems have been developed for PMF including the International Prognostic Scoring System (IPSS) in 200940 and Dynamic IPSS (DIPSS) in 2010.41 IPSS and DIPSS comprised primarily clinical and laboratory parameters, whereas more recent models (reviewed later) have integrated molecular data. The type of MPN phenotypic driver mutation has been shown to have prognostic significance in MF. Specifically, a CALR type-1 mutation has been shown to be a good prognostic factor in several recent MF prognostic scoring systems.42-44 Historically, “triple negative” MF was considered to have a negative prognosis. but this was not confirmed in a recent study.39 The presence of concomitant mutations has been found to have prognostic significance in MF both with respect to the presence of specific mutations17 and the number of mutations.16 These findings were confirmed recently, where HMR patients in both prefibrotic-MF and overt-MF had a lower OS and a lower leukemia-free survival, and the negative outcome was worsened by the presence of multiple mutations.39 Recently, 3 separate prognostic scoring systems were published in MF (Table 1), although none of these separates prefibrotic-MF from overt-MF.
Prognostic scoring models in MF
The Myelofibrosis Secondary to PV and ET-Prognostic model (MYSEC-PM) is a prognostic scoring system specifically designed for patients with MF arising from antecedent PV or ET, which comprises both clinical and molecular data.42 The variables considered are (1) age at diagnosis of secondary MF, (2) presence of anemia, (3) presence of thrombocytopenia, (4) high circulating peripheral blasts, (5) CALR unmutated genotype, and (6) the presence of constitutional symptoms. These variables are used to stratify the patients into 4 risk groups: low, intermediate-1, intermediate-2, and high risk. Notably, other nonphenotypic driver mutations (ie, somatic mutations in genes other than JAK2, CALR, or MPL) are not included in MYSEC-PM. As in the case of primary MF, it is suggested that intermediate-2 and high-risk patients be considered for allogeneic bone marrow transplantation. MYSEC-PM is the first prognostic scoring system dedicated specifically to post-PV and post-ET MF, is uncomplicated, and the data required for calculating the score are readily available in the clinic. MYSEC-PM is also available in the form of an online calculator for determining the score (http://www.mysec-pm.eu/).
The Mutation-Enhanced International Prognostic Score System (MIPSS-70) and MIPSS-70 plus are new prognostic scoring systems dedicated to transplantation-age patients (<70 years).43 MIPSS-70 plus includes cytogenetic information, whereas MIPSS-70 does not. The MIPSS-70 includes the parameters (1) presence of anemia, (2) leukocytosis or leukopenia, (3) thrombocytopenia, (4) circulating blasts, (5) constitutional symptoms, (6) bone marrow fibrosis grade, (7) IPSS/DIPSS-plus category, (8) MPN phenotypic driver mutations, (9) absence of a CALR type 1–like mutation, (10) individual HMR mutations, (11) HMR category, (12) and presence of ≥2 HMR mutations. Each parameter has a weight of 1 point, except for the leukocytosis, thrombocytopenia, and presence of ≥2 HMR mutations, which are each weighted 2 points. The patients are then stratified into 3 categories: low, intermediate, or high risk. The strength of this scoring system is the comprehensive integration of clinical, laboratory, and molecular data. The scoring system is quite complex, but it exists on an online platform with a user-friendly interface (http://www.mipss70score.it/). The goal of this prognostic score is to better select patients as candidates for allogeneic stem cell transplantation.
The Genetically Inspired Prognostic Scoring System (GIPSS) is a prognostic scoring system based only on the molecular features of the patient.44 It considers (1) very high-risk karyotype, (2) unfavorable karyotype, (3) absence of CALR-type 1–like mutation, and (4) presence of SRSF2 mutation, ASXL1 mutation, or U2AF 1Q157 mutation. It defines 4 risk categories: low, intermediate-1, intermediate-2, and high. The novel aspects are the use of molecular information only, the stratification of karyotype, and the substitution in the HMR mutations of U2AF 1Q157 for IDH1/2. This scoring system focuses on a detailed molecular stratification of patients and does not incorporate any clinical parameters. As a result, it may be more useful in early-stage MF patients in which it has the potential to predict outcomes in the absence of clinical signs of progressive disease. It is likely to be less useful in patients with advanced MF, in which the management and prognosis are typically independent of the molecular status of the patient. A suggested role is in the early identification of candidates for consideration for allogeneic stem cell transplant.
Although these retrospective studies and comparison with more widely used prognostic scoring systems (eg, DIPPS) seem to indicate specific advantages for each of the 3 new prognostic models described previously, further validation in prospective clinical trials will be required before these new models can be fully incorporated by physicians into their clinical decision making.
What is the significance of molecular genetics for MPN treatment?
JAK2 inhibitors
Following the discovery of the JAK2 V617F mutation in 2005, JAK2 inhibitors were developed as rationally designed targeted therapy in MPN.45 The oral JAK1/2 inhibitor, ruxolitinib, has received approval from the US Food and Drug Administration for the treatment of intermediate- or high-risk MF on the basis of the results of the Controlled Myelofibrosis Study with Oral JAK Inhibitor Treatment-I (COMFORT-I)46 and COMFORT-II47 clinical trials and for the treatment of patients with PV who are refractory to or intolerant of hydroxyurea on the basis of the results of the Randomized Study of Efficacy and Safety in Polycythemia Vera With JAK Inhibitor INCB018424 Versus Best Supportive Care (RESPONSE)48 clinical trial. Although ruxolitinib has clinical efficacy in MF, it is not strongly clonally selective for MPN cells harboring either JAK2V617F or CALR mutations; as a result, mutant allele burdens do not decrease substantially on treatment. When the 4-year data were reported from the COMFORT-I study, a phase 3 trial in MF, only 12% of patients showed a >50% decrease in JAK2V617F allele burden and <2% patients achieved complete molecular remission (CMR).49 In a retrospective analysis of CALR-mutant patients treated with ruxolitinib on the COMFORT-II study (another phase 3 trial in MF), there was no significant change in the CALR-mutant allele burden after a median of 60 weeks of treatment with ruxolitinib, despite CALR-mutant patients demonstrating comparable clinical responses to JAK2-mutant patients.50 In the RESPONSE study, a phase 3 trial in PV, the mean percent reduction in JAK2V617F allele burden was 40% at 4 years,51 suggesting that ruxolitinib may have stronger clonal selectivity in PV compared with MF. The presence of concomitant mutations has been shown to affect clinical response to ruxolitinib in MF, where patients with ≥3 mutations were less likely to achieve a spleen response and had a shorter time to treatment discontinuation.52 ASXL1 mutations in particular have been associated with worse outcomes in patients with MF treated with ruxolitinib, as discussed previously.29,30 Despite the limitations of ruxolitinib and despite the fact that only a minority of patients with MF completed 5 years of treatment with ruxolitinib on the COMFORT-II study, the Kaplan-Meier estimated probability of survival at 5 years was 56% with ruxolitinib and 44% with best available therapy, suggesting that ruxolitinib may prolong survival in MF.53 In fact, in a retrospective analysis of the COMFORT-II study, the Kaplan-Meier estimated probability of survival indicated a survival benefit even in the HMR group, suggesting that some of the benefit of ruxolitinib in MF may not be due to anticlonal effects.54 With the identification of the crystal structure of the pseudokinase domain of JAK2,55 the potential to develop JAK2V617F mutant-specific inhibitors has been advanced, which should allow for more potent on-target inhibition without the myelosuppressive effects resulting from inhibition of wild-type JAK2 in normal cells.
Interferon
Interferon has a long history in the treatment of MPN,56 and it has been recognized that a minority of interferon-treated PV and ET patients achieve CMR.57-59 A correlation between attaining CMR and achieving a durable remission has been reported.60 One possible mechanism by which interferon achieves molecular remissions in MPN may relate to its ability to stimulate normally quiescent stem cell populations into cycle. Because this cycling is more marked in JAK2V617F-positive HSC, interferon leads to preferential depletion of MPN stem cells in Jak2V617F mouse models,61,62 a finding that has also been reported in primary MPN samples.63 The presence of concomitant mutations (eg, in epigenetic regulators such as TET2, DNMT3A) has been associated with molecular resistance to interferon,64 and more recent studies indicate this is also the case in patients with MF treated with interferon.65,66
Back to the MPN case
The patient’s DIPSS and DIPPS-plus scores both indicate low-risk category. The MIPSS70 score indicates an intermediate-risk category, with a 5-year OS estimated at 67%. The MIPSS70-plus score indicates a low-risk category, with a 5-year OS estimated at 100%. The GIPSS score indicates an intermediate-1 risk category. Because the patient is not symptomatic from his enlarged spleen and does not have substantial constitutional symptoms, treatment with ruxolitinib is not believed to be indicated at this time. In view of the thrombocytosis and concomitant risk factors for cardiovascular disease, aspirin 81 mg daily is initiated. Given that the patient is eligible for allogenic stem cell transplant and given the presence of a HMR mutation (ie, ASXL1), HLA typing of the patient’s siblings is performed; however, allogeneic transplant is not recommended at this time (MIPSS70-plus score is low risk).
Concluding remarks
The cooccurrence of a good prognostic mutation (eg, CALR) with an HMR mutation (eg, ASXL1) in MF can present a challenge to physicians making treatment decisions. The development of prognostic models that integrate molecular variables with clinical parameters has enabled more accurate risk stratification of patients with MF and allowed physicians to develop evidence-based personalized treatment approaches. In the coming years, our expectation is that molecular genetics will be used increasingly to risk-stratify patients with PV and ET. Ultimately, to advance the treatment of MPN will require the use of molecular genetic information not just for risk stratification, but also for therapeutic intervention. In this regard, the development of biologically based treatments rationally designed to target specific molecular vulnerabilities (eg, MPN disease–initiating mutations such as CALR) have the potential to be transformative in MPN, particularly if used early in the course of the disease.
Acknowledgments
This work was supported by National Institutes of Health grant R01HL131835 (A.M.), a Damon Runyon clinical investigator award (A.M.), and the MPN Research Foundation (A.M.). A.M. is a Scholar of The Leukemia & Lymphoma Society. M.C. has received salary support from Novartis. The authors thank Will Duke (Mullally Laboratory) for generating the figures and table.
Correspondence
Ann Mullally, Harvard Institutes of Medicine Building, Rm 738, 77 Ave Louis Pasteur, Boston, MA 02115; e-mail: amullally@partners.org.