
Abstract
Recent advances in genomics have greatly expanded the spectrum of primary immune deficiencies (PIDs). Along with the identification of pathogenic variants in novel genes, distinct phenotypes have been associated with different variants in the same gene. Although PIDs have been historically defined based on increased susceptibility to infections, immune dysregulation has emerged as a frequent and in some cases, predominant phenotype. Autoimmune cytopenias with onset in childhood, lasting longer than 12 months, and affecting multiple lineages should raise the suspicion of a possible PID with monogenic origin. Characterization of the various molecular and cellular mechanisms responsible for these unusual manifestations of PIDs, although at times resource intensive, may allow for targeted intervention in many of them.
Learning Objectives
Recognize immune dysregulation as a possible phenotype associated with primary immune deficiencies leading to hematological manifestations and describe the main underlying cellular and molecular mechanisms
Identify potential targeted therapeutic interventions that may be used in primary immune deficiencies associated with immune dysregulation and describe current outcome with such approaches
Clinical case
A 6-year-old male child presented with pallor. Laboratory tests revealed anemia, hemoglobin of 7.7 g/dL, and positive direct antiglobulin test. Lymphadenopathy and splenomegaly were present. His early childhood history was notable for recurrent respiratory tract infections and eczema. He was treated with steroids, which were tapered off successfully. Autoimmune lymphoproliferative syndrome (ALPS) was ruled out based on molecular analysis. Two years later, he developed headache and generalized seizures. Brain magnetic resonance imaging revealed multiple contrast-enhancing lesions. A brain biopsy showed extensive mixed inflammatory cell infiltrates of meninges and brain parenchyma. He responded to a prolonged course of steroids. At 10 years of age, he developed persistent diarrhea and weight loss. Colonoscopy led to a diagnosis of lymphocytic colitis. At 11 years old, lung nodules were demonstrated at chest computed tomography, and splenomegaly persisted. His father had a history of recurrent pneumonias, enteropathy, and severe cytopenias that required splenectomy at young adult age.
Introduction
Since its inception in the 1980s, the classification of primary immune deficiencies (PIDs) has listed a growing number of disorders. In particular, the last classification, compiled by an expert committee of the International Union of Immunological Societies, includes ∼350 forms of PID.1 This expansion has been made possible by advances in genomics and the availability of high-throughput whole-exome sequencing (WES) and whole-genome sequencing (WGS) techniques. Thus far, >330 genes have been identified with pathogenic variants that are associated with PID; furthermore, different variants in the same PID-associated gene may lead to quite distinct clinical and immunological phenotypes either because of different effects on protein expression and function as exemplified by loss-of-function and gain-of-function (GOF) mutations or because of quantitative effects of mutations that may variably perturb immune system development and function. Importantly, appreciation of the natural clinical history of PID has also evolved during the years. Historically, PIDs had been defined based on increased susceptibility to infections. However, immune dysregulation, manifesting with autoimmunity, inflammatory complications, lymphoproliferation, or increased risk of malignancies, is frequently present, and it may even represent the most prominent feature requiring clinical attention (Figure 1). In a recent manuscript surveying 2183 PID patients reported to the French Registry, 571 subjects (26.2%) were found to have autoimmune or inflammatory manifestations.2 Cytopenias in particular were common, accounting for 31.4% of all forms of immune dysregulation, with a relative risk of developing autoimmune hemolytic anemia that was 830-fold higher in PID patients than in the general population. In another single-center study of 80 patients with pediatric Evans syndrome, pathogenic or potentially damaging genetic variants in immune genes were identified in 52 patients, suggesting that occurrence of early-onset multilineage autoimmune cytopenia should prompt genetic testing.3 Other than cytopenia, other autoimmune, lymphoproliferative, and inflammatory manifestations (especially when occurring at pediatric age) may also be indicative of an underlying immune disorder with a genetic underpinning.2 Autoimmune cytopenias, granulomatous lesions, and lymphocytic infiltrates in target organs (like lungs, liver, and gastrointestinal tract) represent a frequent finding in common variable immune deficiency (CVID), and they may even mark the clinical onset of disease.4 Because CVID is a rather common form of PID, it also accounts for the largest number of PID patients manifesting with immune dysregulation. However, when comparing the incidence of autoimmune and inflammatory complications within each group of PID, combined immune deficiency (CID) has emerged as the PID category with the highest incidence of immune dysregulation.2 Here, we will review the main groups of PID associated with immune dysregulation, with a special focus on chronic/recurrent and treatment-refractory cytopenias owing to peripheral destruction, and how to manage these complications (Figure 2). We will also describe how characterization of the underlying molecular and cellular defects has led to development of targeted therapeutic interventions.

Distribution of gene defects included in the last International Union of Immunological Societies (IUIS) Classification of PIDs (1) and causing diseases manifesting predominantly with infections, immune dysregulation, or both: a personal view.
Distribution of gene defects included in the last International Union of Immunological Societies (IUIS) Classification of PIDs (1) and causing diseases manifesting predominantly with infections, immune dysregulation, or both: a personal view.
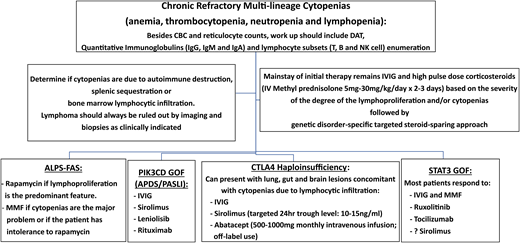
Schematic diagram of a practical approach to the management of cytopenias in immune dysregulatory disorders. APDS, activated phosphatidylinositol 3-kinase δ syndrome; CBC, complete blood count; DAT, direct antiglobulin test; GOF, gain-of-function; IgG, immunoglobulin G; IV, intravenous; MMF, mycophenolate mofetil; NK, natural killer; PASLI, p110 delta-activating mutation causing senescent T cells, lymphadenopathy, and immunodeficiency; PIK3CD, phosphatidylnositol 3-kinase C delta.
Schematic diagram of a practical approach to the management of cytopenias in immune dysregulatory disorders. APDS, activated phosphatidylinositol 3-kinase δ syndrome; CBC, complete blood count; DAT, direct antiglobulin test; GOF, gain-of-function; IgG, immunoglobulin G; IV, intravenous; MMF, mycophenolate mofetil; NK, natural killer; PASLI, p110 delta-activating mutation causing senescent T cells, lymphadenopathy, and immunodeficiency; PIK3CD, phosphatidylnositol 3-kinase C delta.
From ALPS to ALPS-like disorders
ALPS includes a heterogeneous group of disorders characterized by defects of the extrinsic or intrinsic pathways of apoptosis.5 In the majority of the patients, the disease is owing to germline or somatic mutations in the TNFRSF6 gene encoding for the death receptor FS-7–associated surface antigen (FAS) expressed on the surface of activated lymphocytes. Interaction of FAS with FAS ligand (FASLG) triggers a cascade that ultimately results in activation of caspase-8 and -10 and induction of activation-induced cell death (AICD). By contrast, the intrinsic pathway of apoptosis (also known as activated cell autonomous cell death [ACAD]) is induced in response to cytokine withdrawal or on exposure to genotoxic agents. Typically, termination of antigen-induced lymphocyte activation causes an arrest of production of cytokines, including interleukin-2 (IL-2). In turn, this leads to decreased expression of intracellular survival factors (such as BCL-2) and activation of proapoptotic molecules that induce release of cytochrome c from the mitochondria and apoptosome activation. Mutations in FAS, FASLG, and the downstream signaling molecule FADD are a cause of ALPS owing to defects of AICD, whereas somatic mutations of NRAS and KRAS are examples of ALPS-like proliferative disorders associated with monocytosis owing to defects of ACAD.6,7 In these disorders, constitutive activation of the RAS/mitogen-activated protein kinase kinase/extracellular signal-regulated kinase pathway causes phosphorylation and subsequent degradation of BCL2-interacting protein, impeding release of cytochrome c from mitochondria. For this reason, these disorders are also known as RAS-associated autoimmune leukoproliferative disease (RALD).
From a clinical standpoint, defects of both extrinsic and intrinsic pathways of apoptosis cause lymphoproliferation (with splenomegaly and lymphadenopathy), hypergammaglobulinemia, multilineage autoimmune cytopenias (which are often refractory to conventional treatment), and susceptibility to B-cell lymphomas.8 Patients with genetic defects of AICD often have an elevated number of CD4− CD8− (double-negative [DN]) T-cell receptor-αβ+ (TCRαβ+) T cells as well as increased levels of vitamin B12, IL-10, and soluble FASLG.9 Some patients have ALPS owing to somatic mutations of the FAS gene limited to DN T cells; if not properly recognized, this condition may lead to inappropriate therapies.10 However, patients with RALD lack these abnormalities but often present with monocytosis, and they have an increased tendency to develop leukemia with features resembling juvenile myelomonocytic leukemia.11
In addition to somatic mutations in NRAS and KRAS, in recent years a growing number of germline gene defects have been identified by means of WES and WGS in patients who present ALPS-like manifestations but are not mutated in FAS, FASLG, or FADD genes. In particular, an ALPS-like phenotype may associate with CTLA4 and lipopolysaccharide (LPS)-responsive and beige-like anchor protein (LRBA) gene defects as well as activated phosphatidylinositol 3-kinase δ syndrome (APDS), also known as p110d-activating mutation causing senescent T cells, lymphadenopathy, and immunodeficiency (PASLI) syndrome. Furthermore, cytopenias and other autoimmune manifestations are a prominent feature of immune dysregulation-polyendocrinopathy-enteropathy–X-linked (IPEX) syndrome and IPEX-like diseases. Finally, hypomorphic mutations in genes associated with severe combined immune deficiency (SCID) or CID may also be a cause of immune dysregulation. Here, we will review the main clinical, molecular, and cellular features of these immune dysregulation disorders and illustrate how understanding their pathophysiology may lead to a targeted approach to treatment.
ALPS-like disorders
Monoallelic germline mutations in CTLA4, leading to haploinsufficiency, may cause a variable clinical phenotype ranging from multisystem autoimmune manifestations to asymptomatic status.12-14 Variable combinations of hypogammaglobulinemia, recurrent infections, autoimmunity, and lymphoproliferation (presenting with splenomegaly, lymphadenopathy, and lymphocytic infiltration of target organs, such as brain, lungs, and intestine) are the key manifestations. CTLA-4 is a homodimeric inhibitory receptor that binds CD80 and CD86. It plays a critical role in maintaining immune system homeostasis by suppressing T-lymphocyte activation and proliferation, promoting regulatory T (Treg) function, and modulating dendritic cell:T-cell interaction.14 The most frequent presentation in childhood or adolescence is isolated or multilineage cytopenias, often associated with splenomegaly.12,13 In a subset of adult patients, progressive bone marrow hypoplasia (often associated with T-cell infiltrates) is observed, placing CTLA-4 deficiency among the genetic determinants of aplastic anemia.15 Although most patients may have already required corticosteroids, intravenous immunoglobulins, or rituximab for the management of cytopenias by the time a genetic diagnosis is achieved, off-label use of soluble CTLA-4–immunoglobulin fusion protein (abatacept) either alone or in combination with mammalian target of rapamycin (mTOR) inhibitors has been shown to be beneficial in the management of cytopenias and other immunodysregulatory manifestations.16 Safety and efficacy of abatacept for treating chronic cytopenias in CTLA-4 haploinsufficiency are currently being explored in a phase 1/2 randomized, double-blind, placebo-controlled study (NCT03733067).
Biallelic pathogenic variants in the LRBA gene affect surface recycling of the CTLA-4 molecule, causing lysosomal degradation of this molecule.17 Therefore, LRBA deficiency shares several clinical manifestations with CTLA-4 haploinsufficiency, and the same targeted treatment approach has been used for both disorders.17
Heterozygous pathogenic variants in the phosphatidylinositol 3-kinase C delta (PIK3CD) and PIK3R1genes, encoding for p110δ and p85α subunits of the phosphatidylinositol 3-kinase δ (PI3Kδ) enzyme complex, and resulting in increased activity of the PI3K-AKT-mTOR pathway, account for APDS1 and APDS2, respectively.18-20 The clinical manifestations of APDS/PASLI include multilineage cytopenias, lymphoproliferation (with splenomegaly, lymphadenopathy, and nodular lymphoid hyperplasia of the respiratory tract and gastrointestinal mucosa), recurrent infections, and increased susceptibility to B-cell lymphoma.18-20 From an immunological standpoint, patients have a decreased number of naïve T cells, accumulation of senescent T cells, variable degrees of hypogammaglobulinemia often associated with elevated serum immunoglobulin M (IgM), increased number of transitional B cells, reduced proportion of switched memory B cells and impaired response to immunizations.18-20 Use of leniolisib (CDZ173), a novel, potent, and selective oral PI3Kδ inhibitor, in six APDS/PASLI patients in a 12-week, open-label, multicenter, within-subject dose escalation study was recently reported.21 Leniolisib was safe and well tolerated, and it led to a dose-dependent reduction in PI3K/AKT pathway activity and improved the immune dysregulation with normalization of circulating transitional and naïve B cells and reduction in PD-1+ CD4+ and senescent CD57+ CD8+ T cells. Elevated serum IgM and other biomarkers, including interferon-γ, tumor necrosis factor-α, CXCL13, and CXCL10, normalized or decreased with leniolisib. Cytopenias and lymphoproliferation improved in all patients, with index lymph node sizes and spleen volumes reduced by 39% and 40%, respectively. Long-term follow-up safety and efficacy data are available in all six patients who have continued treatment with leniolisib in an extension study at 70 mg twice daily.22 These six patients have been treated for longer than 3 years without experiencing significant adverse events. Three patients have normalized B-cell function and no longer require immunoglobulin supplementation, whereas their cytopenias, including anemia, thrombocytopenia, neutropenia, and lymphopenia, have continued to improve. This experience supports long-term inhibition of p110δ protein as a new targeted therapeutic approach in APDS/PASLI and other disorders of nonmalignant lymphoproliferation associated with the hyperactive PI3K pathway.
IPEX and IPEX-like disorders
IPEX is an X-linked disease characterized by severe autoimmunity, and it is caused by mutations of the FOXP3 gene that controls Treg-cell function.18 In most cases, patients present in the first months of life with a triad of enteropathy, eczema, and type 1 diabetes (T1D). However, other autoimmune manifestations are also possible, especially later in childhood, including cytopenia, hepatitis, nephritis, myopathies, and autoimmune central nervous system disease. Laboratory evaluation of patients with IPEX often reveals lack of circulating CD4+ FOXP3+ T cells; however, CD4+ cells expressing other markers of Treg cells (CD25hi CD127low CTLA-4+ cells) may be present in normal or even increased number, but they lack suppressive activity. If untreated, IPEX is often fatal early in life, and severe organ damage is frequently observed in patients who survive beyond childhood. Medical therapy is based on immunosuppression. Sirolimus is an mTOR inhibitor that has a rather selective effect on effector T cells while sparing Treg cells; for this reason, its use is particularly attractive in patients with IPEX. Although B-cell ablative treatment with rituximab has been often used in patients with cytopenias, most of the organ damage in IPEX is mediated by self-reactive T cells; in such cases, other T-cell immunosuppressive agents, such as mycophenolate mofetil (MMF) and tacrolimus, should be considered other than sirolimus. Definitive cure can be obtained with hematopoietic stem cell transplantation (HSCT), but mixed chimerism complicated by progressive loss of the graft owing to inadequate conditioning may result in relapse of the clinical symptoms.23 In a recent series, long-term survival was similar in patients treated with HSCT compared with those treated with immunosuppression only; however, improved outcome is observed when HSCT is performed in patients with less severe clinical course and better control of the disease.23
Similar to what is described in patients with ALPS with an undetermined genetic defect,24 a number of patients with early-onset autoimmune disease and Treg dysfunction but without FOXP3 mutations have been described.25,26 Within this cohort of patients with IPEX-like presentations, several gene defects have been identified that affect Treg function (Tregopathies).25,26 Although IPEX and IPEX-like diseases share several clinical and laboratory features, eosinophilia and elevated serum IgE are more distinctive features of IPEX. Characterization of molecular signals that are important for Treg function has helped define the pathophysiology of IPEX-like disorders. In particular, IL-2 signaling is essential for Treg survival and activation as well as adaptive immune responses. Mutations of the IL2RA and IL2RB genes (that encode for the α- and β-chains of the IL-2 receptor, respectively) result in autosomal recessive IPEX-like disease associated with increased susceptibility to viral infections and cytomegalovirus (CMV) in particular.27-29
Signal transducer and activator of transcription 3 (STAT3) is a transcription factor that is activated in response to various cytokines and growth factors. GOF mutations of the STAT3 gene cause a pleiomorphic disease characterized by autoimmunity (enteropathy, cytopenia, T1D, hypothyroidism, and interstitial lung disease), lymphoproliferation, eczema, increased susceptibility to infections and malignancies, and short stature.30 The disease may occur as an autosomal dominant trait or in sporadic form. Many of the patients have a reduced number of FOXP3+ cells, and Treg suppressive activity is often reduced. Increased STAT3 function dampens STAT5 activation; therefore, it is not surprising that several features of STAT3 GOF disease are also shared by autosomal recessive STAT5B deficiency.26 The cytopenias seen in these patients are often amendable to steroid-sparing immunosuppressive treatments using MMF. However, progressive pulmonary fibrosis and hypoxic lung damage leading to death have been noted in some patients.
Immune dysregulation in CIDs and other forms of PID
CIDs include a heterogeneous group of disorders that perturb (but do not abrogate) T-cell development and function. The proteins encoded by recombinase activating genes RAG1 and RAG2 initiate the process of V(D)J recombination and are, therefore, necessary for the development of T and B lymphocytes. Although null mutations in these genes are associated with T− B− SCID, hypomorphic mutations that allow residual T- and B-cell development are a cause of atypical SCID or delayed-onset CID manifesting predominantly with granuloma and/or autoimmunity.31 The immune dysregulation associated with hypomorphic RAG mutations reflects impairment of central and peripheral T- and B-cell tolerance.32 In a recent study of 63 patients with RAG deficiency associated with immune dysregulation, a history of cytopenia, often affecting multiple lineages, was documented in 84% of the patients, and other manifestations of autoimmunity (including vasculitis, enteropathy, and endocrinopathy) were documented in fewer patients.33 The median age at presentation of the autoimmune cytopenia was around 2 years, and in some cases, it preceded the occurrence of serious infections. Importantly, neither first-line treatment with intravenous immunoglobulins and steroids nor use of various immunosuppressive agents (MMF, cyclosporine A, tacrolimus, and rituximab) were able to induce stable remission, which was achieved only with HSCT. Granulomatous disease, involving the skin or various tissues (lungs, liver, bone, and soft tissues), was present in a quarter of the patients, and in some cases, the age at onset was in late childhood or even adulthood. In most cases, granulomas responded poorly to anti-inflammatory and immunosuppressive drugs but resolved after HSCT. However, the outcome of patients with atypical forms of RAG deficiency associated with immune dysregulation remains problematic, with a significant death rate even after HSCT, perhaps because a correct diagnosis is established too late in the course of the disease.33
Immune dysregulation and atypical clinical and laboratory features have also been reported in patients with hypomorphic mutations in other SCID-associated genes, including IL2RG, IL7R, JAK3, PRKDC, DCLRE1C, and TRAC.34
Immune dysregulation (with cytopenia, vasculitis, organ-specific autoimmunity, and granuloma formation) is also common in CIDs because of genetic defects that alter the strength of TCR signaling.34 As a result of this defect, these patients are often unable to mount robust adaptive responses, and therefore, they are highly susceptible to infections (in particular owing to Herpesviridae and papillomavirus) and Epstein-Barr virus (EBV)–associated lymphoma. At the same time, a reduced TCR signaling strength may also compromise negative selection of self-reactive T cells and generation and function of Treg cells, causing cytopenia and other manifestations of autoimmunity. A particularly interesting case is represented by the occurrence of severe autoimmunity (intractable bullous pemphigoid, colitis, nephropathy, and hemophilia owing to anti-Factor VIII antibody) in 2 siblings who carried a combination of hypomorphic and activating mutations in the ZAP70 gene.35 In all of these disorders, prompt recognition of the genetic defect and treatment with HSCT are necessary to achieve definitive cure.
Heterozygous STAT1 GOF mutations have been initially described as a cause of autosomal-dominant or sporadic chronic mucocutaneous candidiasis (CMC); however, it has been subsequently shown that this disease is associated with a much broader spectrum of clinical manifestations.36 Other than CMC, these patients may develop other superficial dermatophytoses and invasive mold, coccidioides, and histoplasma infections. Recurrent sinopulmonary infections may lead to bronchiectasis. Nontubercular mycobacterial disease and viral infections (especially caused by Herpesviridae, JC virus, and papillomavirus) have been reported in a significant number of patients. Autoimmunity (manifesting with cytopenia, T1D, thyroiditis, alopecia, and vasculitis) is also very common in STAT1 GOF disease. Nonimmune manifestations (aneurysms, esophageal strictures and increased incidence of malignancies, and squamous cell carcinoma in particular) are also part of the phenotype. The molecular and cellular mechanisms underlying the pleiotropic manifestations of the disease remain ill defined. Increased susceptibility to infections may reflect impaired generation of TH17 cells, and increased interferon-mediated signaling may play a role in the immune dysregulation, similar to what is observed in various interferonopathies. Most patients have been managed with medical therapy, including antifungal treatment and other antimicrobial and immunosuppressive drugs as clinically indicated. Use of Janus-associated kinase (JAK) inhibitors (jakinibs) has been associated with clinical improvement in some but not all patients (see below). Attempts to cure the disease with HSCT have been rather unsatisfactory. In a series of 15 patients, survival was only 40%, and graft failure, graft rejection, and transplant-related toxicity were major complications.37
From characterization of disease pathophysiology to targeted therapy
The identification of the molecular mechanisms underlying the diseases described above has often permitted a rational approach to therapy that, in several cases, is based on targeted interventions aimed to interfere with the key mechanisms of disease (Figure 2).
mTOR inhibitors are being used in diseases where increased AKT phosphorylation or abnormal Treg function is the central pathology. For patients with CTLA-4 and LRBA deficiency, use of CTLA-immunoglobulin (abatacept) and/or sirolimus has improved cytopenias and conferred clinical benefit by reducing lymphoproliferative burden in target organs, such as brain, lungs, gastrointestinal tract, liver, and spleen.17
Jakinibs along with IL-6 inhibition represent an attractive option in patients with STAT1 or STAT3 GOF as well as those with interferonopathies.38 However, by dampening immune responses, these drugs also cause increased susceptibility to infections. The simultaneous use of jakinibs and other immunosuppressive agents (and steroids in particular) has been associated with an increased risk of JC virus and BK virus infection or reactivation. Careful monitoring of the virus status (including regular assessment of EBV and CMV viral load) must be part of the monitoring plan.
For patients with APDS/PASLI, a targeted approach to therapy is based on the use of sirolimus or everolimus (to block the mTOR pathway). Moreover, preliminary results have shown promise for the PI3K inhibitor leniolisib as a suitable targeted treatment of patients with APDS1,21 and a double-blind controlled confirmatory study is currently under way for both APDS1 and APDS2 patients (NCT02859727). However, additional long-term safety and efficacy studies are necessary to assess the role of targeted therapeutics as single agents and in combination vis-a-vis HSCT in some PIDs with immune dysregulation.
Evolution of the clinical case
A CTLA4 mutation was suspected, and a heterozygous splice site mutation, causing in-frame deletion of the extracellular domain of CTLA-4, was confirmed by targeted sequencing.
At 13 years of age, the proband developed new brain lesions, which were treated with a combination of steroids and sirolimus, leading to quick resolution. The patient has remained symptom free on sirolimus treatment since. The proband’s father, who also developed progressive lung involvement, was also diagnosed with CTLA-4 haploinsufficiency and stabilized with sirolimus. However, his enteropathy and intestinal inflammation only responded to intravenous abatacept.
Acknowledgment
This work was supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Correspondence
Luigi D. Notarangelo, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Building 10, Room 5-3950, 10 Center Dr, MSC 1456, Bethesda, MD 20892; e-mail: luigi.notarangelo2@nih.gov.
