Abstract
The short telomere syndromes encompass a spectrum of clinical manifestations that present from infancy to late adulthood. They are caused by mutations in telomerase and other telomere maintenance genes and have a predominantly degenerative phenotype characterized by organ failure across multiple systems. They are collectively one of the most common inherited bone marrow failure syndromes; however, their most prevalent presentations are extrahematopoietic. This review focuses on these common nonhematologic complications, including pulmonary fibrosis, liver pathology, and immunodeficiency. The short telomere syndrome diagnosis informs clinical care, especially in guiding diagnostic evaluations as well as in the solid organ transplant setting. Early recognition allows an individualized approach to screening and management. This review illustrates a myriad of extrahematopoietic presentations of short telomere syndromes and how they impact clinical decisions.
Learning Objectives
Describe the common presentations of extrahematopoietic short telomere disease
Identify implications for diagnostic and management decisions
Introduction
Telomere length is genetically determined with a discrete normal range.1,2 Germline mutations in telomerase and other telomere maintenance genes also accelerate the telomere shortening that normally occurs with aging and result in a spectrum of premature aging syndromes marked by degenerative disease. There is also a specific cancer risk that includes most commonly the marrow-derived malignancies, myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML), with squamous cell cancers being more rare.3,4 Studies in mice have shown that telomerase itself is not essential and that the short telomere length, which is inherited with genetic anticipation, is the primary determinant of phenotype severity.5,6 Dyskeratosis congenita, characterized by a triad of mucocutaneous manifestations, including reticular skin pigmentation abnormalities, nail dystrophy, and mucosal leukoplakia, was the first recognized telomere disease in humans.7,8 Bone marrow failure is a common complication, with a significant percentage of patients with classic dyskeratosis congenita developing at least a single-lineage cytopenia by the age of 30 years,9,10 and often evolving into variable degrees of aplastic anemia.11 Other rarer variants of infant-onset disease have recently been reviewed elsewhere.12 Adult-onset disease is marked by pulmonary fibrosis; given the known prevalence of this lung disease and the percentage of patients with germline telomere defects, it is estimated to account for ≥90% of presentations.13
The pediatric and adult telomere phenotypes of bone marrow failure and pulmonary fibrosis have recently been found to share germline defects in telomere maintenance.14,15 For this review, we refer to them as the “short telomere syndromes” to both indicate their underlying molecular defect and signify the frequent co-occurring syndromic manifestations.2 This term also distinguishes this spectrum from the hypothesized group of “long telomere syndromes” that are thought to manifest as a familial clustering of cancer.16,17 This review focuses on the nonhematologic features of the short telomere syndromes, which can precede the onset of clinically apparent hematologic disease, and their significance for patient care. The case-based approach highlights how the diagnosis of a short telomere syndrome impacts clinical management with a focus on the most common extrahematopoietic complications, including pulmonary fibrosis, liver pathology, and immunodeficiency.
Case 1
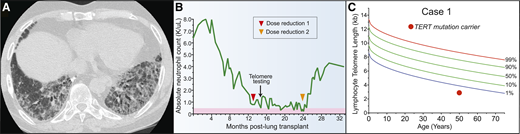
A 51-year-old woman presented to clinic with neutropenia 12 months after lung transplant for the diagnosis of idiopathic pulmonary fibrosis (IPF) (Figure 1A). She reported having a brother who was recently diagnosed with IPF. Her laboratory evaluation was remarkable for a white blood cell count of 2.0 × 103 cells/μL with an absolute neutrophil count of 800 cells/μL, an absolute lymphocyte tcount of 600 cells/μL, and a hemoglobin concentration of 11.3 g/dL. Her antirejection medication, mycophenolic acid, was discontinued without improvement in her neutrophil count (Figure 1B). Consultation with hematology did not reveal reversible or infectious causes, and bone marrow evaluation showed a hypocellular bone marrow for age. The co-occurrence of IPF with marrow hypoplasia prompted an evaluation for a telomere disorder. The patient’s telomere length was found to be below the first age-adjusted percentile, and genetic testing identified a pathogenic mutation in the telomerase reverse transcriptase gene, TERT (Figure 1C). Because of progressive neutropenia, tacrolimus dosing was also reduced to target lower trough levels. She soon recovered and was able to discontinue granulocyte colony-stimulating factor support (Figure 1B). She continued to have excellent allograft function with no evidence of rejection at 36 months after transplant.

Pulmonary fibrosis is the most common short telomere manifestation. (A) Typical appearance of telomere-related IPF characterized on CT by honeycombing in the lung bases. (B) Schematic showing progressive neutropenia after lung transplant that improved after immunosuppressant regimen was attenuated. (C) Lymphocyte telomere length by flow cytometry and fluorescence in situ hybridization (flowFISH) in representative case.
Pulmonary fibrosis is the most common short telomere manifestation. (A) Typical appearance of telomere-related IPF characterized on CT by honeycombing in the lung bases. (B) Schematic showing progressive neutropenia after lung transplant that improved after immunosuppressant regimen was attenuated. (C) Lymphocyte telomere length by flow cytometry and fluorescence in situ hybridization (flowFISH) in representative case.
Genetics of the short telomere syndromes
Mutations in TERT are the most common cause of the short telomere syndromes, accounting for at least 40% of the cases.3,4 Thirteen other genes have been linked to the short telomere syndromes. These genes are part of the telomerase complex itself, have roles in telomerase RNA biogenesis, telomerase recruitment or telomere protection, or are involved in other telomere maintenance functions. A contemporary list of genes and their roles in telomere length maintenance has recently been reviewed.18 Approximately 20% to 30% of patients with classic short telomere phenotypes do not have an identifiable genetic cause of their disease,3,4,19 and, in these cases, the diagnosis is made clinically on the basis of personal and family history along with measurement of telomere length and/or telomerase RNA levels.20
Age-dependent interpretation of telomere length
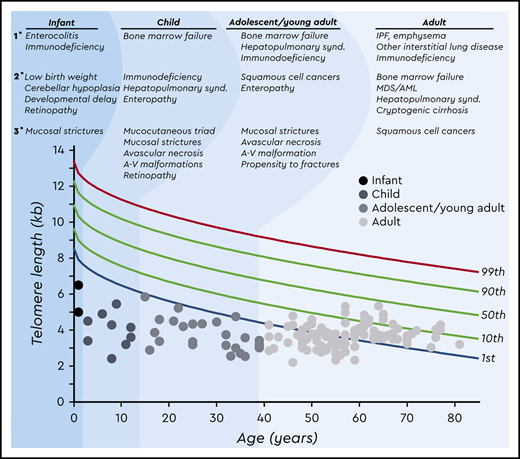
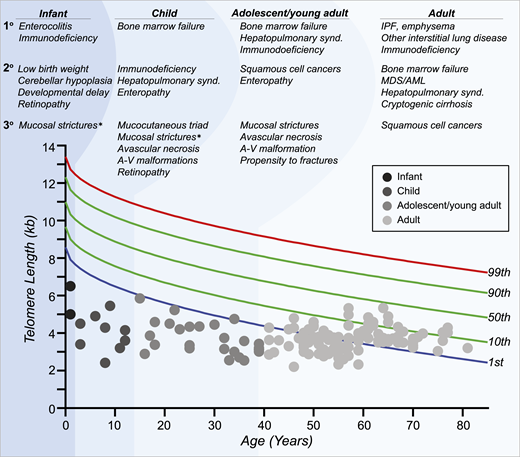
Interpretation of telomere length measurement is age dependent and must take the clinical and, when possible, genetic findings into account. There is not a single threshold applicable to all patients with short telomere syndrome across the age spectrum (Figure 2). Children and adolescents with symptomatic short telomere disease usually fall below the first percentile.21 However, young, often asymptomatic mutation carriers can have telomere lengths between the 1st and 10th percentiles and are at risk for later-onset manifestations (Figure 2).1,22 In those over age 40, telomere length has less diagnostic specificity, and in adults with symptomatic short telomere syndromes of this age, the telomere length can overlap with the lowest decile of the normal population. Furthermore, due to the early mortality of younger patients with short telomeres, nearly all patients over age 60 have telomere lengths above the first percentile.1 In these cases, clinical recognition of the diagnosis based on personal and family history is fundamental along with genetic testing. Other nontelomere inherited bone marrow failure syndromes have also been associated with very short leukocyte telomere lengths.1,23 The biology and natural history of these disorders are distinct from those of the short telomere syndromes, stressing the importance of clinical interpretation of telomere length studies (Figure 2). Finally, lineage-specific telomere length shortening can be seen in some acquired disease states. For example, some patients with autoimmunity have lymphocyte-specific shortening, whereas some patients with MDS acquire granulocyte-restricted shortening.4,24 These observations highlight the importance of measuring telomere length in specific lineages by flow cytometry and fluorescence in situ hybridization (flowFISH).

Age-dependent manifestations of the short telomere syndromes. A schematic telogram demonstrating the typical ranges of telomere length shortening by age of symptomatic disease onset. The threshold for clinically relevant telomere shortening is age and context dependent. Highlighted on top are the predominant manifestations in each of four age groups ordered by prevalence (primary, secondary, and tertiary). Telomere length displays a normal distribution in the population that is defined by the percentile lines labeled on the right. The telomere length is representative here of both total lymphocyte and granulocyte telomere lengths. Each dot represents one patient adapted from symptomatic patients included in Schratz et al.4 *“Mucosal strictures” is a general term that encompasses mucosal defects affecting multiple systems, including lacrimal duct stenosis, esophageal stenosis/strictures/webs, and urethral stenosis/stricture/phimosis. A-V, arteriovenous; synd., syndrome.
Age-dependent manifestations of the short telomere syndromes. A schematic telogram demonstrating the typical ranges of telomere length shortening by age of symptomatic disease onset. The threshold for clinically relevant telomere shortening is age and context dependent. Highlighted on top are the predominant manifestations in each of four age groups ordered by prevalence (primary, secondary, and tertiary). Telomere length displays a normal distribution in the population that is defined by the percentile lines labeled on the right. The telomere length is representative here of both total lymphocyte and granulocyte telomere lengths. Each dot represents one patient adapted from symptomatic patients included in Schratz et al.4 *“Mucosal strictures” is a general term that encompasses mucosal defects affecting multiple systems, including lacrimal duct stenosis, esophageal stenosis/strictures/webs, and urethral stenosis/stricture/phimosis. A-V, arteriovenous; synd., syndrome.
Pulmonary fibrosis is the most common manifestation of the short telomere syndromes
Due to the overall higher prevalence of pulmonary fibrosis in the population, affecting ≥100 000 individuals in the United States alone, and the high prevalence of short telomere gene mutations in those patients, lung disease is the most common manifestation of the short telomere syndromes.13 As in case 1, short telomere syndromes are the most commonly identified genetic cause for familial forms of IPF, accounting for up to 30% to 35% of familial cases.15,20,25-31 In addition to IPF, short telomeres can lead to a broad spectrum of lung disease pathologies that share a progressive course.32 These include nonspecific interstitial pneumonitis, bronchiolitis obliterans organizing pneumonitis, chronic hypersensitivity pneumonitis, pleuroparenchymal fibroelastosis, and emphysema.15,31-34 Studies in mice have linked telomere dysfunction to alveolar stem cell senescence and a resultant epithelial proliferative defect that, with additional injury such as cigarette smoke or pulmonary toxic exposures, is thought to provoke lung remodeling and progressive respiratory disease.35,36 Furthermore, studies in mice and humans suggest the primary short telomere–mediated lung phenotype in some individuals may be influenced by sex differences as well as environmental effects.27,34 For example, within families with short telomere syndromes, pulmonary fibrosis predominates in never smokers, whereas smokers, especially females, are at risk for developing young-onset emphysema alone or in combination with pulmonary fibrosis.34
IPF, characterized radiographically by traction bronchiectasis and reticular honeycombing preferentially affecting the bases and periphery,37 is the most common lung phenotype, accounting for 65% to 70% of lung disease presentations in adults with telomerase mutations.33 Dyspnea and cough are the most common presenting symptoms of pulmonary fibrosis in the short telomere syndromes (Table 1). The average age of onset is in the sixth decade, and symptomatic disease is rare prior to the third decade.27 Younger-onset lung disease can be precipitated by toxic medications previously used in the setting of ablative hematopoietic stem cell transplant.38-40 Pulmonary fibrosis also precedes or co-occurs with telomere-mediated MDS/AML, complicating their treatment, and lung disease is the predominant contributor to mortality in these adult patients with MDS/AML.4 In the short telomere syndromes, the diagnosis of pulmonary fibrosis can be made on the basis of clinical assessment and radiographic studies alone. Molecular and genetic testing for telomere abnormalities has also been suggested as an adjunct to the diagnostic evaluation of IPF due to the high morbidity and risk of mortality with open lung biopsy and the fact that the genetic diagnosis is predictive of the clinical course.32,33
Hematologic manifestations complicate lung transplant for telomere-mediated lung disease
There are currently no medical treatments that have been shown to reverse the extrahematopoietic diseases in the short telomere syndromes and the standard of care for end stage disease is organ transplantation.32 IPF is the leading indication for lung transplant in North America underscoring a particular relevance for the short telomere syndromes.41 The case presented highlights the vulnerable bone marrow reserves in short telomere syndrome patients with pulmonary disease that may be unmasked in the post-transplant setting.42 Telomerase mutation carriers have higher rates of cytopenias, as well as opportunistic infections, particularly cytomegalovirus, and possibly renal insufficiency.42,43 They invariably require dose reduction of the standard anti-rejection regimen, particularly antimetabolite medications.42 Establishing the diagnosis of an underlying short telomere syndrome prior to lung transplant is critical so post-transplant morbidities can be anticipated and possibly averted.42 Apart from attenuation of immunosuppression and use of less myelosuppressive antimicrobial prophylaxis, the primary treatment modalities for post-lung transplant cytopenias are supportive, including transfusions and granulocyte colony‐stimulating factor for severe neutropenia. The benefit of the androgen danazol in this setting has not been studied and its use is controversial due to uncertainties regarding its safety profile in adults44 and concerns it may promote clonal selection in these patients at risk for MDS/AML.22 Management of cytopenias in this setting can be complicated and may require consultation with an experienced center. Going forward, as more patients with telomere-mediated lung disease are identified during the pre-transplant assessment, an inter-disciplinary team that includes hematologists and infectious disease specialists will be important for managing these patients.
Case 2
A previously healthy 57-year-old man presented to his physician for evaluation of unilateral vision loss. His family history was positive for acute leukemia in two siblings. His dilated fundus examination showed opacified lesions tracking centrifugally with the retinal vessels in the right eye with areas of necrosis and hemorrhage (Figure 3A). This was suspicious for cytomegalovirus (CMV) retinitis, and laboratory evaluation demonstrated lymphopenia with an absolute lymphocyte count of 800 cells/μL, absolute CD4 count of 339/μL (normal for age, 500 to 1400/μL), and macrocytic anemia (hemoglobin, 11 g/dL; mean corpuscular volume, 101 fL) but an otherwise intact complete blood count. The result of the patient’s serum CMV polymerase chain reaction assay was positive at 79 000 IU/mL. The result of his HIV antibody test was negative. Treatment of CMV retinitis was initiated with intravitreal foscarnet and systemic ganciclovir. During follow-up care, computed tomography (CT) of the chest, performed for complaints of chronic exertional dyspnea, revealed interstitial lung disease (Figure 3B). These findings prompted an evaluation for a telomere disorder, which demonstrated telomere length below the first age-adjusted percentile (Figure 3C). Genetic evaluation showed no mutations in the known genes. The patient’s hypoxia progressed, and he developed acute-on-chronic respiratory failure. Evaluation identified Pneumocystis jirovecii pneumonia as the cause (Figure 3B). He died 4 months after the diagnosis of his CMV retinitis despite maximum supportive care.
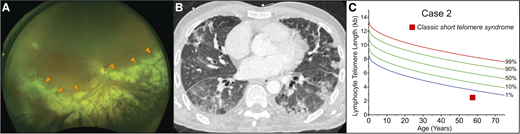
Infectious complications can precede onset of bone marrow failure in the short telomere syndromes. (A) Wide-field fundus photograph of CMV retinitis with intraretinal whitening (orange arrowheads) and necrosis along the retinal vasculature and progressing centrifugally. Image generously provided by Dr. Yonwook Kim of the Boston University Medical Center Department of Ophthalmology. (B) Chest CT image showing pleura-based patchy ground-glass opacities and interlobular septal thickening in a patient who developed P. jirovecii pneumonia in the background of interstitial lung disease and CMV retinitis that was ultimately fatal. (C) Lymphocyte telomere length by flowFISH in a patient with telomere-mediated primary T-cell immunodeficiency manifesting in fatal opportunistic infections.
Infectious complications can precede onset of bone marrow failure in the short telomere syndromes. (A) Wide-field fundus photograph of CMV retinitis with intraretinal whitening (orange arrowheads) and necrosis along the retinal vasculature and progressing centrifugally. Image generously provided by Dr. Yonwook Kim of the Boston University Medical Center Department of Ophthalmology. (B) Chest CT image showing pleura-based patchy ground-glass opacities and interlobular septal thickening in a patient who developed P. jirovecii pneumonia in the background of interstitial lung disease and CMV retinitis that was ultimately fatal. (C) Lymphocyte telomere length by flowFISH in a patient with telomere-mediated primary T-cell immunodeficiency manifesting in fatal opportunistic infections.
Short telomere–mediated T-cell immunodeficiency underlies propensity for CMV infection
CMV is a double-stranded DNA virus in the Herpesviridae family that causes opportunistic infections in individuals with T-cell immunodeficiency.45 Patients with short telomere syndromes have been found to have a specific risk for herpes virus infections, with mortality from CMV infection reported in children and adults46,47 (Table 1). In adults, CMV infection manifests often in immunosuppressed patients with short telomeres after lung transplant.43 Lung transplant recipients with a history of IPF and telomere lengths shorter than the age-adjusted 10th percentile, regardless of germline mutation status, have been reported to have a 5-fold increased risk of relapsing CMV viremia and potentially fatal CMV end-organ disease.42,43 This predilection for CMV disease is due to both global and CMV-specific T-cell immunodeficiency.43,47 Management of CMV in lung transplant recipients with short telomeres is complicated by drug resistance, as well as cytopenias related to myelosuppressive antiviral medications, such as older-generation medications valganciclovir and ganciclovir. Given these considerations, prevention is paramount, and our practice is to avoid high-risk CMV mismatch (seronegative recipient/seropositive donor) when possible, attenuate immunosuppression, and use CMV prophylaxis after transplant. Consultation with infectious disease specialists is helpful in identifying nonmyelosuppressive state-of-the-art strategies to prevent and manage CMV complications in at-risk patients.
Immunodeficiency as a presenting complication
As in the patient in case 2, outside of the lung transplant setting, opportunistic infection with absent or mild bone marrow failure can be the first presenting feature of an inherited short telomere syndrome.47,48 Children with a rare and severe form of short telomere syndromes including Hoyeraal-Hreidarsson syndrome,49,50 present in infancy with noninfectious enterocolitis due to a primarily B-cell immunodeficiency that is also compounded by telomere-related defects in the intestinal epithelium.51 Outside of this early childhood period, opportunistic infections due to the T-cell immunodeficiency predominate and, in addition to CMV end-organ disease, may also present as herpes zoster reactivation, Candida esophagitis, and P. jirovecii pneumonia, for example (Table 1).47,52,53 Due to short telomere-induced apoptosis and secondary depletion of T-cell precursors, children and adults with short telomere syndromes have low T-cell counts in addition to profound functional defects that include depleted naive T-cell pools and a restricted T-cell repertoire.47 Laboratory evaluation of lymphocyte subsets and immunoglobulin levels at diagnosis may be helpful to identify those at highest risk of infection and for consideration for stem cell transplant if the patient is eligible or antimicrobial prophylaxis when the patient is not. T-cell immunodeficiency is an underrecognized indication for stem cell transplant in the absence of bone marrow failure in some patients.47 Recognizing those with a short telomere–mediated immune defect is critical because these individuals require attenuated transplant protocols.54 Patients with unrecognized short telomere syndromes who receive immunosuppression for “idiopathic” disease often develop severe opportunistic infections, underscoring the importance of a high index of suspicion for the diagnosis.
Case 3
A 62-year-old-man presented to clinic with dyspnea; he had been followed for a history of moderate thrombocytopenia unresponsive to immunosuppression. He had been diagnosed with liver cirrhosis 7 years prior and reported going gray at age 23. His family history was negative for lung or hematologic disease. His physical examination showed an age-appropriate man who was otherwise well but had digital clubbing (Figure 4A). Review of recent imaging showed a nodular liver contour, splenomegaly with evidence of varices (Figure 4B), and incidentally detected lower lobe predominant subpleural reticular opacities and traction bronchiectasis. The constellation of bone marrow failure, liver disease, and early signs of pulmonary fibrosis suggested a short telomere syndrome. The patient’s telomere length fell between the 1st and 10th percentile for age, and genetic testing identified a heterozygous frameshift mutation in RTEL1 (Figure 4C). Pulmonary function tests showed a markedly decreased carbon monoxide diffusion capacity with only a mild restrictive ventilatory defect. Agitated saline echocardiography showed delayed right-to-left shunting indicative of intrapulmonary shunting and supportive of the diagnosis of hepatopulmonary syndrome (HPS). The patient’s immunosuppression was withheld, given the diagnosis of telomere-mediated bone marrow failure, and he is being monitored by a multidisciplinary team for progression of his lung, liver, and marrow disease.
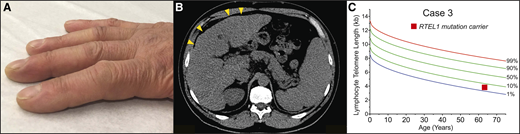
Clubbing and dyspnea can be a presenting feature of telomere-mediated hepatopulmonary syndrome. (A) Image of digital clubbing typically seen. (B) Abdominal CT image showing liver with nodular edges (yellow arrowheads) and secondary splenomegaly due to portal hypertension. (C) The lymphocyte telomere length by flowFISH in older patients with short telomere syndrome presenting over age 60 overlaps with the lower decile of the normal range.
Clubbing and dyspnea can be a presenting feature of telomere-mediated hepatopulmonary syndrome. (A) Image of digital clubbing typically seen. (B) Abdominal CT image showing liver with nodular edges (yellow arrowheads) and secondary splenomegaly due to portal hypertension. (C) The lymphocyte telomere length by flowFISH in older patients with short telomere syndrome presenting over age 60 overlaps with the lower decile of the normal range.
Liver and lung disease co-occur with marrow failure in patients with telomere maintenance defects
Hepatic pathologies have been reported in ∼10% of patients with short telomere syndromes,55 and the triad of bone marrow failure, liver disease, and pulmonary fibrosis has been proposed to be defining for adult-onset disease.14 Early reports identified cryptogenic cirrhosis in patients with dyskeratosis congenita and IPF.55,56 Mouse studies suggested short telomeres decreased regenerative capacity and increased susceptibility to liver injury, resulting in hepatic steatosis and fibrosis with repeated insults.57 Recent studies in patients have also linked the short telomere defect to noncirrhotic portal hypertension.58 This liver disease predominantly manifests as HPS and is characterized by arterial hypoxemia with evidence of intrapulmonary vascular dilatation and secondary arteriovenous connections, both in the lung and in the abdomen.58 As in case 3, the diagnosis of HPS is suspected in patients with short telomere syndrome with progressive dyspnea and a low carbon monoxide diffusion capacity on pulmonary function studies out of proportion to the extent of parenchymal lung disease and spirometry defects. Carbon monoxide diffusion capacity is also a useful, noninvasive method to monitor for disease progression, but it may be difficult to interpret in patients with lung parenchymal disease. Digital clubbing is generally identifiable by physical examination. Mild to moderate splenomegaly may be evident, and hypersplenism can be a significant contributor to thrombocytopenia and neutropenia; this pathology should also be considered in the differential diagnosis for delayed reconstitution or progressive cytopenias after allogeneic stem cell transplant for telomere-mediated marrow failure or immunodeficiency. The diagnosis of HPS can be made by transthoracic agitated saline echocardiogram. The stigmata of underlying liver disease may be subtle, and the diagnosis requires a high index of suspicion. With progression, the liver contour can become increasingly nodular with compensatory hypertrophy of the caudate lobe, which can appear similar to cirrhotic liver disease, but cirrhosis in young patients may not be detected. Histopathologic findings are often nonspecific and may be interpreted as nodular regenerative hyperplasia, among other pathologies.55,58 In contrast to typical cirrhotic liver disease, liver synthetic function is generally well preserved. Patients with short telomere syndrome with HPS share a progressive natural history that has been well documented with complications due to severe hypoxia and portal hypertension, including gastrointestinal bleeding. The median time to death or liver transplant is 6 years from the onset of dyspnea symptoms, with a range of 4 to 10 years.58 Liver transplant reverses the hypoxic defect in patients with telomere-mediated HPS; transplant recipients, however, remain at risk for progression of other short telomere manifestations, including pulmonary fibrosis.58
Clinical and diagnostic screening for extrahematopoietic manifestations
The physical examination with an informed index of suspicion for these age-dependent extrahematopoietic complications should drive the care of patients with short telomere syndromes. Because of the age-dependent natural history of the telomere disorders, there is no one-size-fits-all approach to screening and surveillance. As shown in Table 2, in the Johns Hopkins Telomere Center, we assess patient symptoms and rely on the physical examination to guide testing targeted at the complications highlighted in Table 1. We also factor in telomere length to anticipate the onset of symptoms and to individualize a program for surveillance following the data from large patient series.1,4 We are judicious about diagnostic testing for lung and liver disease in the absence of symptoms, given the high risk of morbidity of interventions, the risk of radiation exposure, and the low yield in unselected age groups. Because the risk of parenchymal pulmonary disease increases with age but is rare prior to age 40, we discuss the risks and benefits of a high-resolution computed tomography in asymptomatic adults older than 40. The advantage of early detection in this group is to facilitate early referral to pulmonary clinics because some cases of pulmonary fibrosis in patients with short telomeres are known to have a rapidly progressive course. Early treatment, however, has not been shown to change the disease course, and the general natural history of short telomere syndromes is slowly progressive.
Summary
A subset of patients with short telomere syndromes can be clinically identified on the basis of personal or family history of hematologic, lung, and liver disease. In other cases, a high index of suspicion in certain settings, such as idiopathic disease of the bone marrow, immune system, lung, or liver, may be necessary to prompt testing. As consideration for an inherited syndrome is now standard for aplastic anemia management, this diagnosis should also be considered in some patients with extrahematopoietic presentations, including IPF, especially in those undergoing lung transplant evaluations, those with primary immunodeficiencies, and patients with liver disease, particularly those with HPS. The diagnosis of a short telomere syndrome informs additional screening and diagnostic evaluations and has critical implications for management. As attenuated approaches to stem cell transplant for aplastic anemia have improved short-term outcomes, efforts are evolving for developing similar approaches to lung and liver transplantation.
Acknowledgments
The author is grateful to Mary Armanios for helpful discussions and comments on the manuscript and figures. The author thanks all the patients, families, and their referring physicians who have contributed to the Johns Hopkins Telomere Syndromes study. This work was supported by a grant from the National Institutes of Health, National Heart, Lung, and Blood Institute (T32HL007525), the Turock Scholars Fund to the Johns Hopkins Telomere Center, and the MacMillan Pathway to Independence Award at the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins.
Correspondence
Kristen E. Schratz, Johns Hopkins University School of Medicine, 1800 Orleans St, Bloomberg 11379, Baltimore, MD 21287; e-mail: kschrat1@jhmi.edu.