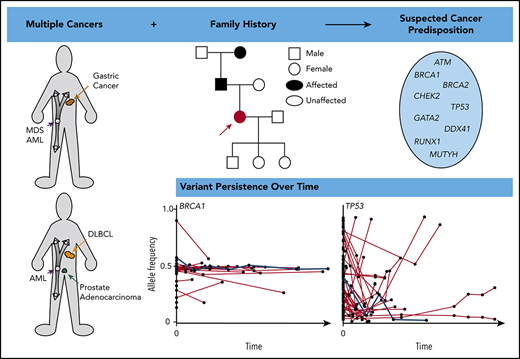
Abstract
Next-generation sequencing (NGS) of bone marrow and peripheral blood increasingly guides clinical care in hematological malignancies. NGS data may help to identify single nucleotide variants, insertions/deletions, copy number variations, and translocations at a single time point, and repeated NGS testing allows tracking of dynamic changes in variants during the course of a patient’s disease. Tumor cells used for NGS may contain germline, somatic, and clonal hematopoietic DNA alterations, and distinguishing the etiology of a variant may be challenging. We describe an approach using patient history, individual variant characteristics, and sequential NGS assays to identify potential germline variants. Our current criteria for identifying an individual likely to have a deleterious germline variant include a strong family history or multiple cancers in a single patient, diagnosis of a hematopoietic malignancy at a younger age than seen in the general population, variant allele frequency > 0.3 of a deleterious allele in a known germline predisposition gene, and variant persistence identified on clinical NGS panels, despite a change in disease state. Sequential molecular testing of hematopoietic specimens may provide insight into disease pathology, impact patient and family members’ care, and potentially identify new cancer-predisposing risk alleles. Ideally, individuals should give consent at the time of NGS testing to receive information about potential germline variants and to allow future contact as research advances.
Learning Objectives
Know when to suspect germline cancer predisposition and how it may impact a patient’s disease and clinical course
Understand how clinical next-generation sequencing of tumor cells may be used to identify potential germline cancer-predisposition variants
Comprehend the evolving nature of genomics pertaining to germline predisposition, implications when considering hematopoietic stem cell transplants using related donors, and informed consent for clinical testing of diseased tissue
Clinical case
A 73-year-old Caucasian woman presented with fatigue, and a complete blood cell count revealed a total white cell count of 2400 per microliter, hemoglobin of 11.4 g/dL, and a platelet count of 104 000 per microliter. The patient was referred to a hematologist who took a detailed personal and family history. Her past medical history was notable for gastric cancer that was treated with a partial gastrectomy followed by oxaliplatin/folinic acid/fluorouracil (FOLFOX) chemotherapy. Although 6 cycles were recommended, the patient stopped chemotherapy after the third cycle because of delayed blood cell count recovery. Her family history included a father diagnosed with “some kind of blood cancer” at ∼75 years of age and a paternal grandmother with “head and neck cancer” at ∼60 years of age (Figure 1A). She noted that her grandmother had never smoked cigarettes or drunk alcohol. A bone marrow (BM) biopsy demonstrated a myelodysplastic syndrome (MDS) characterized by refractory cytopenias with multilineage dysplasia, 15% cellularity, and 14% blasts. Cytogenetic analysis showed a normal karyotype, and molecular profiling using a next-generation sequencing (NGS) panel reported 171 unique variants, including 2 deleterious DDX41 variants: p.Asp140Gfs*2 (p.Asp140fs), initially with a variant allele frequency (VAF) of 0.53, and a p.Arg525His variant with a VAF of 0.27 (Figure 1B). The hematologist recognized the DDX41 p.Asp140fs variant as potentially germline, which is most common in the non-Finnish European population, and the p.Arg525His variant as a hotspot acquired mutation in myeloid malignancies in those with deleterious germline DDX41 variants. She referred the patient to a dermatologist for a skin biopsy.
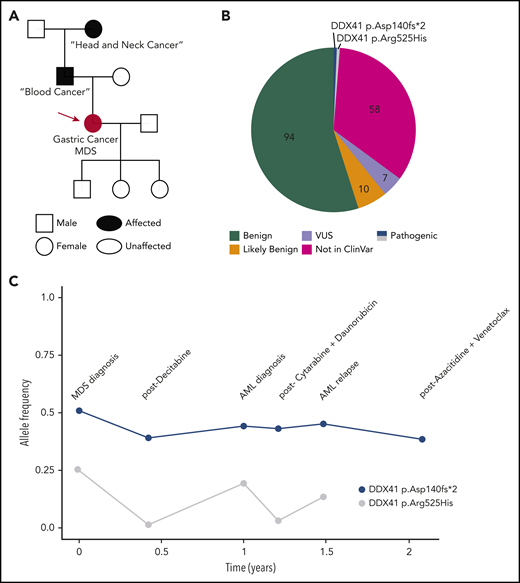
Illustrative case of deleterious DDX41 variants identified during clinical evaluation of a hematopoietic malignancy. (A) Family history revealed 2 cancers in the patient/proband (red circle), a blood cancer of unclear nature in the father, and a head and neck cancer in the paternal grandmother. (B) Molecular profiling via a 150-gene clinical NGS panel identified 171 total variants. After annotation, filtering for clinical relevance, and individual verification by the in-house pathologist, 7 (4%) were reported as variant of uncertain significance, and the 2 (1.1%) DDX41 variants were reported as pathogenic on a final document provided to the treatment team. (C) DDX41 allele VAF is graphed throughout the patient’s clinical course for the 2 variants identified. AML, acute myeloid leukemia.
Illustrative case of deleterious DDX41 variants identified during clinical evaluation of a hematopoietic malignancy. (A) Family history revealed 2 cancers in the patient/proband (red circle), a blood cancer of unclear nature in the father, and a head and neck cancer in the paternal grandmother. (B) Molecular profiling via a 150-gene clinical NGS panel identified 171 total variants. After annotation, filtering for clinical relevance, and individual verification by the in-house pathologist, 7 (4%) were reported as variant of uncertain significance, and the 2 (1.1%) DDX41 variants were reported as pathogenic on a final document provided to the treatment team. (C) DDX41 allele VAF is graphed throughout the patient’s clinical course for the 2 variants identified. AML, acute myeloid leukemia.
Identifying potential germline alterations
Suspicion for germline predisposition may arise from a family history of bleeding, low blood cell counts/function, and/or a personal/family history of cancers. Examples include young age of diagnosis for a specific malignancy, multiple malignancies in the proband, and/or the presence of a hematopoietic or young-onset (<50 years of age) solid tumor within 2 generations of the proband.1 An advanced age at cancer diagnosis does not exclude the potential for a germline-predisposition syndrome. For instance, the average age of diagnosis of a myeloid malignancy in patients with germline DDX41 mutations is ∼70 years.2 Additionally, some inherited predisposition syndromes may be caused by de novo gene mutations (ie, arising during embryogenesis and present in the germline of affected individuals but not the parents), such as GATA2, BRCA1, and BRCA2, among others.3,4
Cultured skin fibroblasts or hair bulbs are considered to the best source for germline DNA.5,6 Skin biopsies may be performed simultaneously with a BM biopsy, when the skin is sterile and anesthetized. Unfortunately, culturing fibroblasts requires several weeks and may be technically challenging, and hair bulbs may be scarce in oncology patients and do not yield large quantities of DNA, limiting downstream testing. In addition to determining the germline status of a particular allele via sequencing from these DNA sources, an allele identified in ≥2 related individuals defines its germline status. Although tempting, peripheral blood (PB) or a lymph node without evidence of tumor as a comparative “normal” sample may confound germline predictions as a result of clonal hematopoiesis (CH) and other rearrangements. Buccal swabs and fingernails may be contaminated with hematopoietic cells.6 Unfortunately, even if the proper material is selected, NGS panels for germline variants differ with regard to which genes are included and the types of variants detected.7 Therefore, identifying a panel that provides comprehensive testing for relevant genes and variant types is essential.
Curation of gene variants is the systematic evidenced-based process by which functional data, population frequency, disease phenotype, familial pedigree, and biologic evidence are integrated to assign clinical significance using the standardized 5-tier system outlined by the American College of Medical Genetics and Genomics and the Association of Molecular Pathology. Tiers include pathogenic, likely pathogenic, variant of uncertain significance, likely benign, or benign.8,9 Formal germline variant deposition occurs in the open access platform ClinVar (ncbi.nlm.nih.gov/clinvar), with curation mediated by expert panels within ClinGen (clinicalgenome.org). Curation rules for germline RUNX1 variants are available from the Myeloid Malignancy Variant Curation Expert Panel (clinicalgenome.org/affiliation/50034/) and are being applied to RUNX1 variants within ClinVar with U.S. Food and Drug Administration recognition.10,11 The ClinGen curation process undergoes reassessment every 2 years to allow updating based on recent literature and revised ClinGen recommendations.
Identifying potential germline alleles from NGS assays of tumor samples
Molecular profiling of specimens containing tumor cells, including tumor/BM biopsies and PB samples, is performed with increasing frequency. All sample types potentially contain germline and somatic variants, and tumor biopsies may also contain PB or infiltrating leukocytes containing independent acquired CH variants within hematopoietic tissue.1 DNA aberrations may arise from several distinct etiologies. Somatic alterations are unique to the tumor itself, CH alterations are derived from the clonal expansion of hematopoietic stem or progenitor cells, and germline variants are present in all nongerm cells in the body (Figure 2). When a population of cells undergoes bulk NGS, each aberration identified is reported with a single VAF, making it difficult to infer the nature of the variant directly. Ideally, germline variants have VAF ∼ 0.5 if heterozygous or VAF ∼ 1.0 if homozygous (Figure 2). However, the VAF must be interpreted relative to germline mosaicism, loss of heterozygosity, copy number variations (CNVs) in tumor cells, insertions/deletions, structural rearrangements, and sequencing artifacts, including statistical fluctuation particularly with shallow sequencing depths. Specific NGS tests (eg, exomes, genomes, targeted gene panels, hot spot panels, and CNV tiling), sequencing technologies, and data processing, alignment, and variant calling/annotation methods may affect the variants identified and the VAF calculated. Therefore, careful interpretation of NGS data from clinical samples is needed. A suggested preliminary screening approach includes determining likely germline vs somatic status considering the gene, VAF, purity, and ploidy and then, among the likely somatic alterations, determining whether it is likely derived from tumor or common CH variants from contaminating hematopoietic cells.
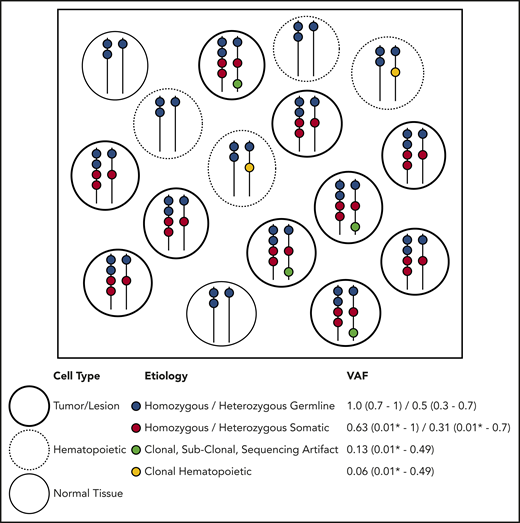
Etiology of DNA alterations in a representative sample of NGS sequencing. The box contains a population of cells (each circle) undergoing bulk NGS. Each color represents a DNA alteration of different etiology. VAF for the representative example is shown with typical ranges observed on clinical NGS panels. This example does not account for CNVs or DNA structural aberrations. *Lower detection limit depends on the depth and platform of NGS.
Etiology of DNA alterations in a representative sample of NGS sequencing. The box contains a population of cells (each circle) undergoing bulk NGS. Each color represents a DNA alteration of different etiology. VAF for the representative example is shown with typical ranges observed on clinical NGS panels. This example does not account for CNVs or DNA structural aberrations. *Lower detection limit depends on the depth and platform of NGS.
If NGS panels include genes that confer germline risk for BM failure or cancer (Table 1), they may provide an opportunity to detect germline variants in patients with hematopoietic malignancies, even when DNA from tumor cells is used.12 For some genes, detection of particular alleles should raise suspicion of germline origin. For example, some germline DDX41 variants are commonly observed in certain populations (opening case), and all truncating variants reported to date are germline.13 For other alleles, such as many RUNX1 or TP53 variants, etiology is difficult to surmise, because the same variant can be somatic or germline. Importantly, suspicion of a germline variant should be confirmed by testing true germline DNA.
Many studies examine the frequency of germline variants identified in tumor-based sequencing and provide a conservative updated list of genes in which germline variants drive tumorigenesis (Table 1).12,14-16 Germline variants are identified on ∼7% to 25% of tumor-only panels, but most are not deleterious.12,14-16 The most frequent genes with germline variants in solid tumors and hematopoietic malignancies include ATM, BRCA1, BRCA2, CHEK2, DDX41, GATA2, MUTYH, and TP53.12,14,16
Some bioinformatic algorithms may suggest germline etiology using single NGS assays.17,18 However, additional insight is gained by examining tests over time. This is illustrated by a second case that highlights an individual with a personal history of diffuse large B-cell lymphoma, prostate cancer, and acute myeloid leukemia (AML), as well as a strong family history of cancer, who was found to have a BRCA1 deletion with VAF ∼ 50% on multiple NGS assays that ultimately was shown to be germline (Figure 3). The patient’s AML had acquired JAK2 and TP53 variants, potentially as a consequence of underlying germline predisposition and environmental risk factors.19 A TSC2 variant was seen after allogeneic hematopoietic stem cell transplantation (HSCT). As this case highlights, a germline variant typically remains detectable over time with a relatively consistent VAF (Figure 4, blue lines), whereas the VAF of somatic variants is prone to change with disease status (Figure 4, red lines). Some gene mutations, like those in TP53, occur most commonly as somatic alleles (Figure 4A, red line). Rarely, individuals who may not have met clinical criteria for Li-Fraumeni syndrome are found to have germline TP53 mutations by tracking the VAF over time or in multiple assays (Figure 4A, blue lines). In our experience, deleterious BRCA1/2 variants are more commonly germline alterations (Figure 4B-C). Following VAF over time is an efficient way to identify potential germline variants using data already collected for diagnostic and prognostic purposes. Other conditions that may yield a stable VAF ∼ 50% include persistent disease, CH, CNVs, and loss of heterozygosity. It is important to remember that germline variants can be deleterious, benign, or of uncertain significance, so the germline nature of a variant should not be equated with being pathogenic. Figure 5 shows an algorithm for identifying germline variants and guiding when to send comprehensive testing. At the University of Chicago, we compare new NGS test results from PB and BM for all patients with hematopoietic malignancies with previous data obtained from that individual. In a typical month, we review data from ∼60 to 85 patients with hematopoietic malignancies who have had multiple NGS tests and identify 1 to 3 (1% to 5%) patients with potentially pathogenic germline variants that were not otherwise detected. This information is conveyed to the patient’s primary oncologist who facilitates genetic counseling and, when appropriate, testing to confirm germline status (Figure 5).

A complex clinical case and VAF of deleterious variants seen over time. A 51-year-old white man had an 8 × 10–cm mass that was determined to be diffuse large B-cell lymphoma (DLBCL). He received 6 cycles of rituximab/cyclophosphamide/doxorubicin/vincristine/prednisone (R-CHOP), 2 cycles of etoposide/methylprednisolone/high-dose cytarabine/cisplatin (ESHAP), and radiation to the mediastinum, ultimately achieving a complete response. At age 57, a screening prostate-specific antigen (PSA) was 12.4 ng/mL. Prostate biopsy showed a 4 + 4 = 8 Gleason score adenocarcinoma, and the patient had a prostatectomy with normalization of his PSA. At age 61 years, he was diagnosed with essential thrombocytosis, with JAK2 p.Val617Phe. He eventually progressed to AML, when a detailed family history was obtained. (A) Family history revealed numerous relatives with cancer: mother, breast cancer (55 years old); father, prostate cancer (69 years old); maternal aunt, ovarian cancer (37 years old); maternal cousin, unknown type of leukemia; paternal grandmother, uterine cancer; paternal grandfather, head and neck cancer; paternal aunt, breast cancer (70 years old); paternal cousin, brain tumor (75 years old); paternal cousin, lymphoma (70 years old); paternal cousin, unknown type of leukemia (12 years old). Molecular profiling at AML diagnosis showed a complex karyotype, including deletions of the long arms of chromosomes 5 and 7. NGS of predominantly leukemia cells from a BM biopsy showed a TP53 mutation and a deletion within BRCA1. The patient underwent induction chemotherapy, and molecular profiling at clinical remission demonstrated persistence of the BRCA1 deletion and loss of the TP53 mutation. Germline genetic testing on DNA derived from the patient’s cultured skin fibroblasts confirmed a germline BRCA1 deletion. He underwent an allogeneic HSCT using an unrelated donor, given the potential risk of the familial BRCA1 deletion, which had been found in an HLA-matched sibling. (B) The VAF of DNA alterations are plotted over time and show persistence of the germline BRCA1 deletion at a relatively high VAF prior to HSCT; the acquired clonal JAK2 and TP53 variants prior to HSCT; and an acquired TSC2 variant post-HSCT of donor origin. Lessons from this case include: (1) The patient was diagnosed with 3 cancers by the time germline testing was performed: DLBCL, prostate cancer, and AML. Genetic counseling and testing were warranted at the time of his first cancer based on his extensive family cancer history. (2) BRCA1 and BRCA2 are Fanconi anemia-like genes,37 encoding proteins important for DNA repair pathways active in the BM. Individuals with BRCA pathway mutations are at increased risk for the development of hematopoietic malignancies.38 In fact, cancer predisposition syndromes generally thought of as predisposing to solid tumors also increase the risk for hematopoietic malignancies, such as Lynch and Li-Fraumeni syndromes.39,40
A complex clinical case and VAF of deleterious variants seen over time. A 51-year-old white man had an 8 × 10–cm mass that was determined to be diffuse large B-cell lymphoma (DLBCL). He received 6 cycles of rituximab/cyclophosphamide/doxorubicin/vincristine/prednisone (R-CHOP), 2 cycles of etoposide/methylprednisolone/high-dose cytarabine/cisplatin (ESHAP), and radiation to the mediastinum, ultimately achieving a complete response. At age 57, a screening prostate-specific antigen (PSA) was 12.4 ng/mL. Prostate biopsy showed a 4 + 4 = 8 Gleason score adenocarcinoma, and the patient had a prostatectomy with normalization of his PSA. At age 61 years, he was diagnosed with essential thrombocytosis, with JAK2 p.Val617Phe. He eventually progressed to AML, when a detailed family history was obtained. (A) Family history revealed numerous relatives with cancer: mother, breast cancer (55 years old); father, prostate cancer (69 years old); maternal aunt, ovarian cancer (37 years old); maternal cousin, unknown type of leukemia; paternal grandmother, uterine cancer; paternal grandfather, head and neck cancer; paternal aunt, breast cancer (70 years old); paternal cousin, brain tumor (75 years old); paternal cousin, lymphoma (70 years old); paternal cousin, unknown type of leukemia (12 years old). Molecular profiling at AML diagnosis showed a complex karyotype, including deletions of the long arms of chromosomes 5 and 7. NGS of predominantly leukemia cells from a BM biopsy showed a TP53 mutation and a deletion within BRCA1. The patient underwent induction chemotherapy, and molecular profiling at clinical remission demonstrated persistence of the BRCA1 deletion and loss of the TP53 mutation. Germline genetic testing on DNA derived from the patient’s cultured skin fibroblasts confirmed a germline BRCA1 deletion. He underwent an allogeneic HSCT using an unrelated donor, given the potential risk of the familial BRCA1 deletion, which had been found in an HLA-matched sibling. (B) The VAF of DNA alterations are plotted over time and show persistence of the germline BRCA1 deletion at a relatively high VAF prior to HSCT; the acquired clonal JAK2 and TP53 variants prior to HSCT; and an acquired TSC2 variant post-HSCT of donor origin. Lessons from this case include: (1) The patient was diagnosed with 3 cancers by the time germline testing was performed: DLBCL, prostate cancer, and AML. Genetic counseling and testing were warranted at the time of his first cancer based on his extensive family cancer history. (2) BRCA1 and BRCA2 are Fanconi anemia-like genes,37 encoding proteins important for DNA repair pathways active in the BM. Individuals with BRCA pathway mutations are at increased risk for the development of hematopoietic malignancies.38 In fact, cancer predisposition syndromes generally thought of as predisposing to solid tumors also increase the risk for hematopoietic malignancies, such as Lynch and Li-Fraumeni syndromes.39,40
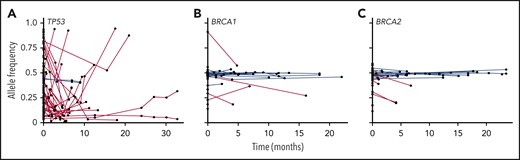
VAFs for TP53, BRCA1, and BRCA2 and over time in patients with hematopoietic malignancies. VAFs of DNA alterations in TP53 (A), BRCA1 (B), and BRCA2 (C) in individual patients at the University of Chicago are graphed over time. Each point indicates an individual variant identified in an in-house NGS assay, and red lines connect likely somatic variants; likely germline variants are shown in blue.
VAFs for TP53, BRCA1, and BRCA2 and over time in patients with hematopoietic malignancies. VAFs of DNA alterations in TP53 (A), BRCA1 (B), and BRCA2 (C) in individual patients at the University of Chicago are graphed over time. Each point indicates an individual variant identified in an in-house NGS assay, and red lines connect likely somatic variants; likely germline variants are shown in blue.
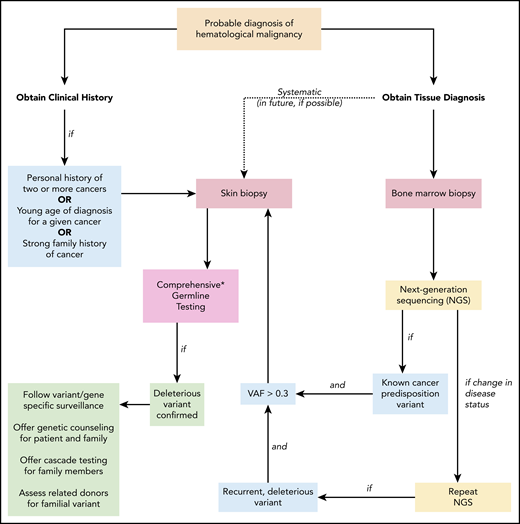
Suggested algorithm for identifying patients with a deleterious germline cancer predisposition variant. When a patient is diagnosed with a hematopoietic malignancy, clinical history and tumor biopsies are performed. Personal history of prior cancer (1 other hematopoietic malignancy or solid tumor, including melanoma in an individual younger than 50 years of age), diagnosis at a younger age than seen in the general population for a given cancer, or a strong family history of cancer (relative diagnosed with cancer within 2 generations of the patient) should prompt a skin biopsy and comprehensive germline testing. If tumor-only NGS identifies a known cancer-predisposition variant and the VAF is > 0.3, germline testing of the variant should follow. As additional NGS tests are performed to monitor the patient’s clinical course, persistent deleterious variants with VAF > 0.3 should prompt consideration of germline status. This is especially warranted if the deleterious variant is present in a gene associated with cancer risk. In the future, systematic collection of a skin biopsy at the time of the initial BM biopsy and culturing of fibroblasts to obtain germline DNA may become standard (dotted line). Once a deleterious germline variant is confirmed, variant/gene-specific surveillance should be followed for the patient (including a risk assessment for cancer involving organs outside the BM), genetic counseling and germline testing should be offered to appropriate family members, and potential risks should be considered if the patient were to undergo related HSCT from a family member sharing the allele. NGS, next-generation sequencing; VAF variant allele frequency. *Comprehensive testing that includes all genes and variant types that confer cancer risk is not standardized and requires careful review of testing options.
Suggested algorithm for identifying patients with a deleterious germline cancer predisposition variant. When a patient is diagnosed with a hematopoietic malignancy, clinical history and tumor biopsies are performed. Personal history of prior cancer (1 other hematopoietic malignancy or solid tumor, including melanoma in an individual younger than 50 years of age), diagnosis at a younger age than seen in the general population for a given cancer, or a strong family history of cancer (relative diagnosed with cancer within 2 generations of the patient) should prompt a skin biopsy and comprehensive germline testing. If tumor-only NGS identifies a known cancer-predisposition variant and the VAF is > 0.3, germline testing of the variant should follow. As additional NGS tests are performed to monitor the patient’s clinical course, persistent deleterious variants with VAF > 0.3 should prompt consideration of germline status. This is especially warranted if the deleterious variant is present in a gene associated with cancer risk. In the future, systematic collection of a skin biopsy at the time of the initial BM biopsy and culturing of fibroblasts to obtain germline DNA may become standard (dotted line). Once a deleterious germline variant is confirmed, variant/gene-specific surveillance should be followed for the patient (including a risk assessment for cancer involving organs outside the BM), genetic counseling and germline testing should be offered to appropriate family members, and potential risks should be considered if the patient were to undergo related HSCT from a family member sharing the allele. NGS, next-generation sequencing; VAF variant allele frequency. *Comprehensive testing that includes all genes and variant types that confer cancer risk is not standardized and requires careful review of testing options.
Testing related HSCT donors
Allogeneic HSCT is often considered for patients with hematopoietic malignancies. Determining whether a germline variant is pathogenic is critical for choosing the donor, because family members are generally preferred.20 When related donors have been used who were found retrospectively to have deleterious germline variants in RUNX1 or CEBPA, poor outcomes are observed, including poor hematopoietic stem cell mobilization by the donor,21,22 failure or delay in engraftment,21,23 poor immune function,21,23 early relapse,23 donor-derived leukemias,22,24,25 and new diagnosis of leukemia in the related donor after stem cell mobilization/collection.24,25 Thus, using related donors with deleterious germline RUNX1 or CEBPA mutations is not advised, but our knowledge about other genes is limited. As research expands to assess the impact of deleterious germline variants in HSCT donors and recipients, we may learn which are permissive for successful transplantation, which are prudent to avoid, and whether mobilization with granulocyte colony-stimulating factor and other agents confers additional risk. Furthermore, matched unrelated donors within close-knit populations, such as the Ashkenazi Jewish population, may also harbor potential pathogenic germline alleles. In fact, all donors, including cord blood units, may have deleterious germline variants, but this potential hazard is not assessed routinely. Currently, there is a range of criteria in clinical practice, with some transplant centers mandating donor testing and rejecting any donor with a deleterious germline variant in any cancer risk gene, whereas others forgo testing altogether. Overall, more data are needed to determine whether there are deleterious variants permissive for HSCT (as highlighted in the second case).
Patient consent
As the clinical utility of NGS sequencing becomes commonplace, it is important to consider the dynamic nature of the field. The number of actionable variants is increasing, and investigators are enhancing ways to use these data.26-28 Personalized medicine trials are underway, and researchers are developing inventive new ways to interpret NGS findings, such as monitoring measurable residual disease, tracking clonal expansion to understand tumor evolution, and identifying novel germline cancer-predisposition syndromes.29-31 Health care providers and patients should understand the limitations, the benefits, and the potential that information may change with advances in NGS and variant interpretation. Some institutions consent to patient understanding that NGS testing may reveal potential germline variants, some give patients the option to receive such data, and other centers forgo consent altogether. Overall, it may be important to discuss the challenges and limitations and to disclose the evolving nature of knowledge with patients as we perform more clinical NGS panels.32,33
Returning to the opening case
Testing DNA from cultured skin fibroblasts confirmed the germline status of the DDX41 p.Asp140fs allele, as suspected. The patient shared this information with her three children, who considered cascade testing. Over the next 2 years, the patient progressed to AML and received multiple chemotherapy regimens. Molecular testing revealed the germline variant repeatedly, with the VAF dropping to 0.39 on 1 occasion (Figure 1C), highlighting variability in technical assay performance. The VAF of the p.Arg525His allele varied widely in parallel with the BM blast percentage (Figure 1C).
Key points include:
The diagnosis of 2 cancers (ie, gastric cancer and MDS in this case) should prompt a skin biopsy at the time of the diagnostic BM biopsy (Figure 5).
Careful selection of NGS panels is required to ensure comprehensive testing. Importantly, DDX41 is missing from many commercial NGS platforms.7 Each assay is unique in design and implementation, resulting in particular technical limitations and variabilities.
Initial descriptions of cancer-predisposition syndromes may be based on small case numbers, and expansion of the phenotype may occur over time. This case raises the question as to whether some solid tumors may occur more frequently in individuals/families with deleterious germline DDX41 variants.
Repeating NGS assays throughout a patient’s disease provides a means to identify potential germline variants. Germline status should be considered when a deleterious variant is identified in a known cancer-predisposition gene with a VAF > 0.3, if the variant is usually of germline nature (opening case) or persists over time (Figures 1C, 3B, and 5, and the second case).
Conclusions
NGS panels of PB, BM, or tumor specimens are used to identify therapeutically actionable aberrations, for risk stratification, and to provide insights into disease biology. Interpretation may be challenging because somatic, CH, or germline findings may be present. If systematic germline testing is not available, a family history or multiple cancer diagnoses, early age of diagnosis, the presence of known germline predisposition gene variants, or persistent DNA alterations on sequential assays over time should raise concern for germline predisposition. Repeating clinical NGS panels may be an opportunity to distinguish variant etiology and should be considered throughout a patient’s clinical course, although this does not replace comprehensive germline testing. Frequent reevaluation of NGS results may be important as research progresses, and germline testing of related HSCT donors should be considered if a patient harbors a pathogenic germline variant. Outcomes after HSCT using donors with germline variants warrant careful study, and discussions at the time of NGS on tumor cells ideally includes the indication of patient preference for disclosure of possible germline alleles.
Acknowledgments
The authors thank Jeremy Segal, George Steinhardt IV, and Nifang Niu (Department of Pathology, The University of Chicago) for ongoing collaboration on the use of panel-based testing of hematopoietic tissues, as well as their patients for continued involvement in research on germline predisposition to hematopoietic malignancies.
Correspondence
Lucy A. Godley, 5841 S. Maryland Ave, MC 2115, Chicago, IL 60637; e-mail: lgodley@medicine.bsd.uchicago.edu.