

Abstract
In amyloid light chain (AL) amyloidosis, a small B-cell clone, most commonly a plasma cell clone, produces monoclonal light chains that exert organ toxicity and deposit in tissue in the form of amyloid fibrils. Organ involvement determines the clinical manifestations, but symptoms are usually recognized late. Patients with disease diagnosed at advanced stages, particularly when heart involvement is present, are at high risk of death within a few months. However, symptoms are always preceded by a detectable monoclonal gammopathy and by elevated biomarkers of organ involvement, and hematologists can screen subjects who have known monoclonal gammopathy for amyloid organ dysfunction and damage, allowing for a presymptomatic diagnosis. Discriminating patients with other forms of amyloidosis is difficult but necessary, and tissue typing with adequate technology available at referral centers, is mandatory to confirm AL amyloidosis. Treatment targets the underlying clone and should be risk adapted to rapidly administer the most effective therapy patients can safely tolerate. In approximately one-fifth of patients, autologous stem cell transplantation can be considered up front or after bortezomib-based conditioning. Bortezomib can improve the depth of response after transplantation and is the backbone of treatment of patients who are not eligible for transplantation. The daratumumab+bortezomib combination is emerging as a novel standard of care in AL amyloidosis. Treatment should be aimed at achieving early and profound hematologic response and organ response in the long term. Close monitoring of hematologic response is vital to shifting nonresponders to rescue treatments. Patients with relapsed/refractory disease are generally treated with immune-modulatory drugs, but daratumumab is also an effective option.
Learning Objectives
Learn how to timely and correctly diagnose light chain amyloidosis
Learn how to use currently available and novel regimens to treat light chain amyloidosis based on accurate risk and response assessment
Clinical case
A 65-year-old man with a history of hypertension developed worsening exertional dyspnea over the course of 6 months. During the previous 6 months he had progressively reduced and eventually discontinued his angiotensin-converting enzyme inhibitors because of “resolution” of hypertension. His cardiologist suspected amyloid heart involvement based on an echocardiography and recommended cardiac magnetic resonance (CMR) imaging, which showed late gadolinium enhancement. 99mTc-hydroxymethylene-diphosphonate scintigraphy revealed cardiac uptake. A diagnosis of transthyretin (ATTR) amyloidosis was presumed. The patient was referred to a medical geneticist to rule out hereditary amyloidoses and to a hematologist to rule out light chain (AL) amyloidosis. Genetic testing for hereditary ATTR amyloidosis was negative. Immunofixation revealed κ Bence Jones protein. The patient was then referred to our center for amyloid typing and presented with New York Heart Association class III (NYHA class III) heart failure and postural hypotension. The κ-free light chain (FLC) concentration was 206 mg/L (ratio [FLCR], 10.3, and differential FLC [dFLC], 186 mg/L); bone marrow plasma cell (PC) infiltrate was 12% without chromosomal abnormalities; blood count, calcium, and liver function test results were normal; estimated glomerular filtration rate (eGFR) was 48 mL/min; proteinuria was 2.8 g per 24 hours, predominantly albumin; N-terminal pronatriuretic peptide type-B (NT-proBNP) was 10 625 ng/L (upper reference limit [url], 227 ng/L); and cardiac troponin I (cTnI) was 124 ng/L (url, 44 ng/L). A computed tomographic (CT) scan showed no bone lesions. Abdominal fat aspirate showed amyloid deposits typed as AL κ by immunoelectron microscopy (IEM). A diagnosis of AL amyloidosis with cardiac (stage IIIb) and renal (stage II) involvement was established. The patient received attenuated treatment with cyclophosphamide, bortezomib, and dexamethasone in subintensive care. Treatment was associated with fluid retention. Nevertheless, he received the second cycle as an outpatient. After 2 cycles, very good partial response (VGPR) was reached (dFLC, 11 mg/L; FLCR, 2.1; and persistence of κ Bence Jones protein), with improvement of markers of cardiac (NT-proBNP, 7225 ng/L) and renal (proteinuria, 1.7 g per 24 hours) involvement. Two more cycles were administered that were accompanied by fluid retention but did not improve hematologic (dFLC, 9 mg/L; FLCR, 2.0; and persistence of κ Bence Jones protein) and organ (NT-proBNP, 6792 ng/L; proteinuria, 1.5 g per 24 hours) response. Heart failure improved (NYHA class II), and treatment was discontinued based on the patient’s preference. Follow-up testing was scheduled every 3 months. After 15 months, markers of organ involvement were stable (NT-proBNP, 7471 ng/L, proteinuria, 1.4 g per 24 hours), but FLC increased (dFLC, 98 mg/L; FLCR, 5.9). The patient was treated with daratumumab, bortezomib, and dexamethasone. After 1 week, VGPR was reestablished (dFLC, 8 mg/L; FLCR, 1.8; and persistence of κ Bence Jones protein), and after 4 months, complete response (CR) was attained (dFLC, 3 mg/L; FLCR. 1.2; and negative serum and urine immunofixation) with cardiac response (NT-proBNP, 2809 ng/L). Twelve months later, CR had been maintained, and minimal residual disease (MRD) was not detectable by next-generation flow cytometry.
Introduction
In systemic AL amyloidosis a PC clone, or, less frequently, a lymphoplasmacytic or marginal zone lymphoma, produces a toxic LC that causes organ dysfunction and damage and forms amyloid fibrils in tissues. In contrast, localized deposition of LCs causes nodules to develop in the skin and in the respiratory, urinary, and gastrointestinal tracts, with local symptoms and a benign course that usually is managed with local treatment.1 In systemic AL amyloidosis, the PC clone is usually small (median infiltrate, 10%), and presents t(11;14) and gain 1(q21) in ∼50% and 20% of clones, respectively, whereas high-risk aberrations are uncommon.2,3 Patients whose PC clones harbor t(11;14) have a worse outcome with bortezomib and immunomodulatory drugs (IMiDs), whereas gain 1(q21) is associated with poorer results with oral melphalan.3-5
Heart involvement is the major determinant of survival. Preclinical models and clinical observation of rapid cardiac improvement after a decline in LC concentration disclosed a direct cardiotoxic effect of the circulating precursor.6,7 The severity of organ involvement is assessed with biomarkers combined in accurate staging systems (Table 1).8-12 Survival also depends on hematologic response (HR), because LCs are the agents directly causing organ dysfunction. If the disease is not treated promptly and effectively, organ dysfunction progresses and eventually leads to death. Recent trials of immunotherapies targeting the amyloid deposits (the anti-fibril antibody NEOD001 and the combination of the amyloid P component [which targets small-molecule miridesap], and dezamizumab) failed. Only one anti-amyloid fibril antibody CAEL-101 is still under evaluation (www.clinicaltrials.gov #NCT04304144). Current treatments target the underlying clone and are aimed at suppressing the production of LCs to restore organ function and extend survival. Advanced organ involvement, particularly cardiac, is associated with early death and causes extreme frailty limiting the delivery of effective therapy. In recent years, advancements in biomarker-based risk stratification and monitoring of response and novel anti-PC agents has improved outcomes. Early and correct diagnosis is the prerequisite to beneficial use of these tools.
Diagnostic workup
The clinical presentation of AL amyloidosis depends on organ involvement, and it is protean and deceitful. Symptoms are often misinterpreted and recognized late. When they appear, organ involvement is often irreversible. However, cardiac and renal amyloidosis can be detected by NT-proBNP and albuminuria before overt heart failure and nephrotic syndrome arise. Moreover, a monoclonal component can be found at least 4 years before diagnosis.13 Therefore, hematologists can intercept, diagnose, and treat patients during the presymptomatic stage, by including biomarkers of organ involvement in the monitoring panel of subjects with monoclonal gammopathy of undetermined significance (MGUS).14 Recent findings suggest that CMR can detect very early cardiac involvement15 ; hence, CMR may be used as a confirmatory test in subjects with MGUS in whom elevated NT-proBNP is found. However, the cost effectiveness of this approach is unknown. A diagnostic flowchart for AL amyloidosis is reported in Figure 1.

Presenting features and diagnostic algorithm for AL amyloidosis. Amyloidosis can be suspected if elevated biomarkers of organ involvement are detected during follow-up of patients with MGUS or if suggestive symptoms arise. The first scenario is ideal and enables early presymptomatic diagnosis. Based on relative rates of progression, appropriate screening programs should detect 1 patient with MGUS progressing to AL amyloidosis for every 7 to 10 who develop multiple myeloma. In patients with MGUS in whom elevated NT-proBNP or BNP is found, cardiac magnetic resonance imaging can be used as a higher specificity confirmatory test. If symptoms of systemic amyloidosis arise in a patient in whom a preexisting monoclonal gammopathy is not known, the first step should be searching for a monoclonal component, particularly if heart involvement is suspected, so as not to delay diagnosis. Only the combination of immunofixation of both serum and urine and FLC measurement grant adequate diagnostic sensitivity to detect amyloidogenic monoclonal proteins. MS-based methods are under investigation. Patients with suspect cardiac amyloidosis without monoclonal components can have an attempted nonbiopsy diagnosis of ATTR amyloidosis with cardiac scintigraphy with bone tracers. Validated tracers are 99mTc-diphosphono-propanodicarboxylic acid, 99mTc-pyrophosphate, and 99mTc-hydroxymethylene diphosphonate. DNA analysis is necessary to differentiate between hereditary and wild-type ATTR amyloidosis and to rule out other rarer hereditary forms. All other patients require a tissue diagnosis. Amyloid deposits can be found in abdominal fat, minor salivary glands, and bone marrow, and most patients can be spared biopsy of the involved organ. However, if amyloidosis is deemed probable, for a prompt start of treatment, organ biopsy should not be deferred. Amyloid deposits are recognized as nonbranching fibrils of 7 to 10 nm in width, detected by light microscopy with green birefringence under polarized light after staining with Congo red or by electron microscopy. The diagnostic sensitivity of abdominal fat aspirate combined with bone marrow or minor salivary gland biopsy is ∼90% at referral centers, but the recognition of amyloid deposits is affected by the experience of the pathologist. With a few exceptions (eg, patients with a monoclonal component and periorbital purpura and/or macroglossia, or combination of amyloid heart and renal involvement with albuminuria), the clinical presentation of AL amyloidosis cannot reliably be differentiated from that of other types of systemic amyloidosis. Thus, amyloid tissue typing with adequate technology is mandatory. Standard light microscopy immunohistochemistry does perform satisfactorily, and patients should be referred to specialized centers for typing with adequate technology (immunohistochemistry with custom-made antibodies, IEM, or MS). Accurate clonal studies, biomarker-based staging, and assessment of comorbidities are necessary to design the therapeutic strategy. MRI, magnetic resonance imaging.
Presenting features and diagnostic algorithm for AL amyloidosis. Amyloidosis can be suspected if elevated biomarkers of organ involvement are detected during follow-up of patients with MGUS or if suggestive symptoms arise. The first scenario is ideal and enables early presymptomatic diagnosis. Based on relative rates of progression, appropriate screening programs should detect 1 patient with MGUS progressing to AL amyloidosis for every 7 to 10 who develop multiple myeloma. In patients with MGUS in whom elevated NT-proBNP or BNP is found, cardiac magnetic resonance imaging can be used as a higher specificity confirmatory test. If symptoms of systemic amyloidosis arise in a patient in whom a preexisting monoclonal gammopathy is not known, the first step should be searching for a monoclonal component, particularly if heart involvement is suspected, so as not to delay diagnosis. Only the combination of immunofixation of both serum and urine and FLC measurement grant adequate diagnostic sensitivity to detect amyloidogenic monoclonal proteins. MS-based methods are under investigation. Patients with suspect cardiac amyloidosis without monoclonal components can have an attempted nonbiopsy diagnosis of ATTR amyloidosis with cardiac scintigraphy with bone tracers. Validated tracers are 99mTc-diphosphono-propanodicarboxylic acid, 99mTc-pyrophosphate, and 99mTc-hydroxymethylene diphosphonate. DNA analysis is necessary to differentiate between hereditary and wild-type ATTR amyloidosis and to rule out other rarer hereditary forms. All other patients require a tissue diagnosis. Amyloid deposits can be found in abdominal fat, minor salivary glands, and bone marrow, and most patients can be spared biopsy of the involved organ. However, if amyloidosis is deemed probable, for a prompt start of treatment, organ biopsy should not be deferred. Amyloid deposits are recognized as nonbranching fibrils of 7 to 10 nm in width, detected by light microscopy with green birefringence under polarized light after staining with Congo red or by electron microscopy. The diagnostic sensitivity of abdominal fat aspirate combined with bone marrow or minor salivary gland biopsy is ∼90% at referral centers, but the recognition of amyloid deposits is affected by the experience of the pathologist. With a few exceptions (eg, patients with a monoclonal component and periorbital purpura and/or macroglossia, or combination of amyloid heart and renal involvement with albuminuria), the clinical presentation of AL amyloidosis cannot reliably be differentiated from that of other types of systemic amyloidosis. Thus, amyloid tissue typing with adequate technology is mandatory. Standard light microscopy immunohistochemistry does perform satisfactorily, and patients should be referred to specialized centers for typing with adequate technology (immunohistochemistry with custom-made antibodies, IEM, or MS). Accurate clonal studies, biomarker-based staging, and assessment of comorbidities are necessary to design the therapeutic strategy. MRI, magnetic resonance imaging.
A tissue biopsy is always necessary to establish the diagnosis of AL amyloidosis. Notably, other types of systemic amyloidosis can have clinical presentations that overlap that of AL amyloidosis (Table 2). Of these, wild-type transthyretin (ATTRwt) amyloidosis, is the most common. Effective treatments are now available for wild-type and hereditary ATTR amyloidosis, but correct amyloid typing is mandatory. The other, rarer types should not be disregarded. In contrast to AL amyloidosis, the diagnosis of ATTRwt amyloidosis requires tissue typing only in patients in whom a monoclonal component is detected. In the absence of a monoclonal component, a nonbiopsy diagnosis of ATTRwt amyloidosis is possible based on cardiac uptake of bisphosphonate scintigraphy tracers.16 Patients with cardiac AL amyloidosis usually have no or modest uptake; however, patients may have intense uptake. Moreover, one-fourth to one-third of patients with ATTRwt have a monoclonal component. AL amyloidosis is by far the most rapidly progressing type of cardiac amyloidosis and is the one that benefits most from early initiation of effective therapy. Thus, the first step in the diagnostic workup of cardiac amyloidosis should be searching for monoclonal components. In the present clinical case, a substantial diagnostic delay resulted from deferred testing for PC dyscrasia.
Once the diagnosis of AL amyloidosis is established, the characteristics of the underlying clone and the extent and severity of organ involvement should be studied (Figure 1). This information is essential for designing the therapeutic strategy.
Up-front therapy
Treatment should be risk adapted, considering the severity of organ involvement, characteristics of the clone, and comorbidities and seeking to deliver the most rapid and effective therapy patients can safely tolerate. Delicate up-front therapy can sometimes trigger early improvement of organ dysfunction, allowing for subsequent, more aggressive treatment. Early and profound reductions of the amyloid LC are associated with the greatest chance of organ improvement, and prolongation of progression-free (PFS) and overall (OS) survival.17-19 Changes in biomarkers can be used to assess response to therapy according to validated criteria (Table 3).12,17,20,21 The optimal end point of therapy is still a matter of debate. Achievement of organ response can indicate that the amyloid LC level has fallen below the concentration needed to sustain organ dysfunction. Organ response can parallel HR, as it did in the patient described herein, but it is sometimes delayed. For this reason, assessment of treatment efficacy and a decision to shift to rescue regimens should be based on early HR assessment (3 months after autologous stem cell transplantation [ASCT], 1 to 2 months after nontransplantation therapies). Achievement of organ response and profound clonal response should be the long-term goal of therapy. Individual frailty, age, bone marrow PC infiltration, residual organ dysfunction, and treatment tolerability should be considered to decide whether CR should be pursued. Novel definitions of deep HR are being investigated.18,19 The relatively low sensitivity and imprecision of available FLC assays demand novel technologies, such as mass spectrometry (MS)–based detection of monoclonal components22 and assessment of MRD23 that await validation in AL amyloidosis.
Accurate risk stratification is crucial in designing the treatment strategy (Figure 2). Approximately 20% of patients with newly diagnosed disease are candidates for ASCT, and more can become eligible after effective up-front therapy. Moreover, pretransplantation induction therapy independently improves PFS.24 In a large series, 84% of patients attained HR after ASCT (VGPR, 33%; CR, 39%),25 and the CR rate can increase to ∼60% (∼40% MRD−) with posttransplantation bortezomib-based treatment.26 OS of patients who reach CR with ASCT is >50% at 15 years.27
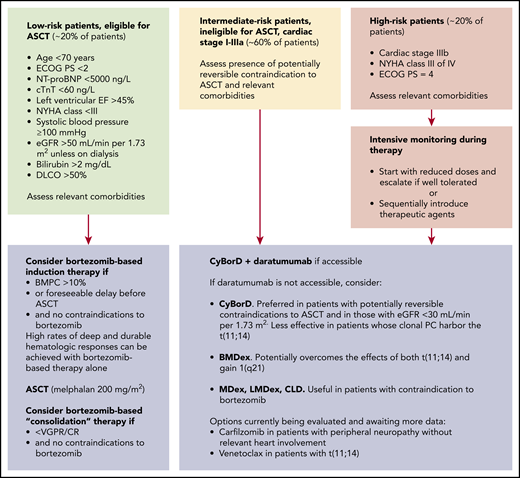
Treatment strategy for patients with newly diagnosed AL amyloidosis. The design of the treatment strategy requires accurate risk stratification. In the past, transplant-related mortality related to advanced amyloid organ involvement was very high. Refinement of selection criteria with the inclusion of cardiac biomarkers resulted in a significant improvement in tolerability of ASCT. Subjects who are not transplantation eligible at diagnosis, may become suitable transplantation candidates if they attain organ response after up-front therapy. Melphalan dose adjustment to extend ASCT eligibility does not decrease toxicity but negatively impacts response rate and should be discouraged. Pretransplant therapy with bortezomib-based regimens is beneficial in patients with a bone marrow PC infiltrate >10% but can be considered in all patients to attain rapid reduction of the amyloid light chain, if harvesting procedures and ASCT scheduling can result in a relevant delay. Moreover, recent data indicate that induction therapy independently increases PFS. Importantly, bortezomib-based therapy alone can grant satisfactory (CR and/or organ response) and durable response in some patients who may then not proceed to ASCT. Posttransplantation therapy with bortezomib-based regimens increases CR rate and extends PFS in patients who attain less than VGPR after ASCT. Amyloidogenic PCs depend on proteasomes to survive the stress caused by toxic LCs, resulting in particular sensitivity to proteasome inhibition, and bortezomib is also the cornerstone of treatment of patients who are not eligible for ASCT. Combination of bortezomib-based regimens with daratumumab will most likely become novel standards of care based on the results of recent clinical trials. In current clinical practice, daratumumab is still not widely accessible, and CyBorD is preferred over BMDex in patients with renal failure and in those who may later become eligible for ASCT, whereas BMDex may overcome the negative effects of both t(11;14) and gain 1q21. Venetoclax is also an appealing option for patients with t(11;14), but few data are available so far. Relevant comorbidities include potential contraindications to bortezomib, such as peripheral neuropathy and pulmonary fibrosis. Oral MDex or immunomodulatory drug (IMiD)-based regimens are valuable alternatives for subjects with contraindications to bortezomib. Carfilzomib can also be considered in patients with peripheral neuropathy, carefully balancing potential cardiac toxicity. Patients with non-PC clones should be treated with regimens specifically targeting the underlying amyloid clone.
Treatment strategy for patients with newly diagnosed AL amyloidosis. The design of the treatment strategy requires accurate risk stratification. In the past, transplant-related mortality related to advanced amyloid organ involvement was very high. Refinement of selection criteria with the inclusion of cardiac biomarkers resulted in a significant improvement in tolerability of ASCT. Subjects who are not transplantation eligible at diagnosis, may become suitable transplantation candidates if they attain organ response after up-front therapy. Melphalan dose adjustment to extend ASCT eligibility does not decrease toxicity but negatively impacts response rate and should be discouraged. Pretransplant therapy with bortezomib-based regimens is beneficial in patients with a bone marrow PC infiltrate >10% but can be considered in all patients to attain rapid reduction of the amyloid light chain, if harvesting procedures and ASCT scheduling can result in a relevant delay. Moreover, recent data indicate that induction therapy independently increases PFS. Importantly, bortezomib-based therapy alone can grant satisfactory (CR and/or organ response) and durable response in some patients who may then not proceed to ASCT. Posttransplantation therapy with bortezomib-based regimens increases CR rate and extends PFS in patients who attain less than VGPR after ASCT. Amyloidogenic PCs depend on proteasomes to survive the stress caused by toxic LCs, resulting in particular sensitivity to proteasome inhibition, and bortezomib is also the cornerstone of treatment of patients who are not eligible for ASCT. Combination of bortezomib-based regimens with daratumumab will most likely become novel standards of care based on the results of recent clinical trials. In current clinical practice, daratumumab is still not widely accessible, and CyBorD is preferred over BMDex in patients with renal failure and in those who may later become eligible for ASCT, whereas BMDex may overcome the negative effects of both t(11;14) and gain 1q21. Venetoclax is also an appealing option for patients with t(11;14), but few data are available so far. Relevant comorbidities include potential contraindications to bortezomib, such as peripheral neuropathy and pulmonary fibrosis. Oral MDex or immunomodulatory drug (IMiD)-based regimens are valuable alternatives for subjects with contraindications to bortezomib. Carfilzomib can also be considered in patients with peripheral neuropathy, carefully balancing potential cardiac toxicity. Patients with non-PC clones should be treated with regimens specifically targeting the underlying amyloid clone.
Most patients with AL amyloidosis are not eligible for ASCT. Oral melphalan + dexamethasone (MDex) has been a standard of care for many years in these subjects. Currently, bortezomib is the backbone of up-front treatment regimens and is combined with MDex (BMDex) or with cyclophosphamide and dexamethasone (CyBorD). A phase 3 trial in intermediate-risk patients (#NCT01277016) showed that BMDex induces a significantly higher HR rate (81% vs 57%; CR, 23% vs 20%; VGPR 42% vs 20%) than MDex, with prolonged OS.28 Cardiac and renal responses were observed in 38% and 44% of cases with BMDex and in 28% and 43% of cases with MDex, respectively.28 In a large, unselected, retrospective series, overall HR rate in response to CyBorD was 65% (CR, 25%; VGPR, 24%), with cardiac response in 33% of patients and renal response in 15%.18 A phase 3 trial comparing CyBorD with CyBorD+subcutaneous daratumumab has been completed (#NCT03201965). The uncontrolled safety assessment performed in a portion of the study showed a remarkable 96% overall HR rate.29 The randomized trial preliminary results indicate that addition of daratumumab results in significantly higher hematologic (92% vs 77%; CR/VGPR, 79% vs 42%), cardiac (42% vs 22%), and renal (54% vs 27%) response rates across cardiac and renal stages and independent of t(11;14) and prolonged PFS.30
Approximately 20% of patients have advanced (stage IIIb) cardiac involvement at diagnosis. Treatment of these patients remains an unmet need. However, if a profound response is reached within 1 month, OS can improve, even in these subjects.31 Ideally, these patients need a very rapidly acting, safe regimen. The safety profile and rapidity of action of subcutaneous daratumumab make this agent appealing in this setting, and a phase 2 trial is under way (#NCT04131309). Supportive therapy is vital to sustaining organ function while chemotherapy is delivered (Figure 3).

Supportive therapy in AL amyloidosis. Supportive measures have a fundamental role in the management of AL amyloidosis, with the goal of improving quality of life, relieving symptoms, and sustaining organ function while anti-PC therapy is delivered and takes effect. The mainstay of supportive treatment is diuretic therapy. However, in amyloidosis, cardiac function is preload dependent, and it is important to avoid reduction of intravascular volume. Angiotensin-converting enzyme inhibitors are generally poorly tolerated because of hypotension: they should be used at the lowest possible dose and discontinued if not well tolerated. Transplantation of the organs involved by amyloidosis may render patients with advanced disease eligible for aggressive specific treatment. The main concerns with organ transplantation are occurrence of amyloidosis in the graft and progression in other organs. However, the availability of effective anti-PC treatments allows for consideration of heart transplantation followed by effective chemotherapy in young patients with isolated severe cardiac involvement. Patients who have advanced, irreversible organ damage, despite achievement of complete HR, can also be considered for transplantation of the organs involved. However, early reports still awaiting confirmation suggest that patients who fail to attain organ response despite having achieved complete HR may have persistent minimal residual clonal disease. In these subjects, further chemotherapy, if deliverable, may lead to minimal residual disease negativity and improvement of organ dysfunction. Implantation of left ventricular assist devices is technically feasible for patients with severe heart failure caused by advanced cardiac amyloidosis, but the possible benefit is unclear.
Supportive therapy in AL amyloidosis. Supportive measures have a fundamental role in the management of AL amyloidosis, with the goal of improving quality of life, relieving symptoms, and sustaining organ function while anti-PC therapy is delivered and takes effect. The mainstay of supportive treatment is diuretic therapy. However, in amyloidosis, cardiac function is preload dependent, and it is important to avoid reduction of intravascular volume. Angiotensin-converting enzyme inhibitors are generally poorly tolerated because of hypotension: they should be used at the lowest possible dose and discontinued if not well tolerated. Transplantation of the organs involved by amyloidosis may render patients with advanced disease eligible for aggressive specific treatment. The main concerns with organ transplantation are occurrence of amyloidosis in the graft and progression in other organs. However, the availability of effective anti-PC treatments allows for consideration of heart transplantation followed by effective chemotherapy in young patients with isolated severe cardiac involvement. Patients who have advanced, irreversible organ damage, despite achievement of complete HR, can also be considered for transplantation of the organs involved. However, early reports still awaiting confirmation suggest that patients who fail to attain organ response despite having achieved complete HR may have persistent minimal residual clonal disease. In these subjects, further chemotherapy, if deliverable, may lead to minimal residual disease negativity and improvement of organ dysfunction. Implantation of left ventricular assist devices is technically feasible for patients with severe heart failure caused by advanced cardiac amyloidosis, but the possible benefit is unclear.
Treatment of relapsed/refractory disease
Patients who do not attain satisfactory response should be shifted to second-line treatment as early as possible. So far, there is no evidence to support maintenance therapy in responders, who should be closely followed. However, there is no consensus on when treatment should be started at relapse. We lack validated hematologic progression criteria, and the definition of PFS varies in different studies. Organ progression criteria predict shorter patient and renal survival and such progression should not be awaited before starting rescue therapy.12,17 In general, organ progression is preceded by FLC increases, which can be small and should not be disregarded.32 Other factors to be considered are FLC level and severity of organ involvement at diagnosis, as well as the quality of response to previous treatment.
The mainstay of rescue therapy is IMiDs that can overcome resistance to alkylating agents and proteasome inhibitors, with an OS benefit in responders. In a pooled analysis of patients enrolled in 2 phase 2 clinical trials of lenalidomide and 1 of pomalidomide, 39% of patients achieved VGPR or CR.33 In a real-world European study of pomalidomide, overall HR rate was 44% (CR, 3%; VGPR, 23%).34 Treatment with IMiDs interferes with cardiac response assessment, being associated with NT-proBNP increase, and worsening renal failure can be observed in patients with proteinuria. A phase 3 study compared the oral proteasome inhibitor ixazomib with physician’s best choice (lenalidomide in 57% of patients) in relapsed/refractory AL amyloidosis.35 The study failed to meet its primary end point, an improvement in overall HR rate (53% vs 51%), but CR rate was higher (26% vs 18%) and PFS longer in ixazomib-treated patients. Ixazomib can be safely combined with lenalidomide and dexamethasone in an all-oral regimen.36
Non-IMiD–based rescue has recently been evaluated. A phase 2 study reported a 57% HR rate (CR 11%) with bendamustine+dexamethasone.37 In the past few months, 2 phase 2 trials and numerous retrospective series have addressed the efficacy of daratumumab-based regimens in relapsed/refractory disease. Sanchorawala et al observed a remarkable 90% response rate (CR 41%) in a phase 2 single-agent trial of daratumumab.38 Cardiac and renal response rates were also high (50% and 67%, respectively).38 In a European trial, 55% of patients responded to daratumumab (CR 8%), with lower cardiac (25%) and renal (31%) response rates.39 Notably, in this trial, >50% of patients had not achieved VGPR or better with previous lines of therapy.39 Close monitoring revealed that most HRs occurred after the first daratumumab infusion.38,39 The largest retrospective study included 168 patients treated with daratumumab alone or combined with bortezomib.40 There was no significant difference in outcome with the 2 regimens, and overall HR rate was ∼65% (CR/VGPR ∼50%).40 Interestingly, shorter PFS was observed in patients with nephrotic syndrome.40 Combination of daratumumab with IMiDs is particularly interesting in relapsed/refractory disease, and several clinical trials are under way.
Conclusion
Despite late recognition of symptoms and delayed referral to a specialized center, the patient described herein benefited from sequential treatment with powerful and rapidly acting regimens guided by biomarker-based risk stratification and monitoring. General practitioners, cardiologists, and nephrologists should be able to recognize symptoms early and to suggest appropriate testing (first, search for a monoclonal component) that can direct patients to the diagnostic pathways of AL or non-AL amyloidosis. Hematologists are in the unique position of recognizing and treating presymptomatic patients. Common pitfalls in the management of patients with AL amyloidosis are reported in Figure 4.

Common pitfalls in the management of patients with AL amyloidosis. A presymptomatic biomarker-based diagnosis is possible in patients at risk (subjects with MGUS and abnormal FLC ratio). Treating patients at early stages facilitates the access to effective therapies and can improve survival. The diagnostic pathways for AL and non-AL amyloidosis are different and the choice depends on the presence or absence of a monoclonal component. AL amyloidosis progresses more rapidly, but available treatments can rapidly reverse the course of the disease. The diagnosis of AL amyloidosis should not be delayed. Positive cardiac scintigraphy with bone tracers is not enough to establish a diagnosis of ATTR amyloidosis in a patient with a monoclonal component. Uncharacterized amyloid deposits on a tissue biopsy in a patient with a monoclonal component are not enough to establish a diagnosis of AL amyloidosis. Prespecified treatment duration and/or number of cycles should be avoided in AL amyloidosis. The goal is rapid and deep HR and if it is not reached, rescue therapy is needed. Organ response can sometimes be delayed.
Common pitfalls in the management of patients with AL amyloidosis. A presymptomatic biomarker-based diagnosis is possible in patients at risk (subjects with MGUS and abnormal FLC ratio). Treating patients at early stages facilitates the access to effective therapies and can improve survival. The diagnostic pathways for AL and non-AL amyloidosis are different and the choice depends on the presence or absence of a monoclonal component. AL amyloidosis progresses more rapidly, but available treatments can rapidly reverse the course of the disease. The diagnosis of AL amyloidosis should not be delayed. Positive cardiac scintigraphy with bone tracers is not enough to establish a diagnosis of ATTR amyloidosis in a patient with a monoclonal component. Uncharacterized amyloid deposits on a tissue biopsy in a patient with a monoclonal component are not enough to establish a diagnosis of AL amyloidosis. Prespecified treatment duration and/or number of cycles should be avoided in AL amyloidosis. The goal is rapid and deep HR and if it is not reached, rescue therapy is needed. Organ response can sometimes be delayed.
After many years without positive controlled studies, in the past few months, 3 phase 3 trials have been published.28,30,35 Daratumumab combined with bortezomib emerged as a new standard of care. Yet, therapeutic innovation left old questions unanswered and opened new ones. A standard of care for high-risk patients is lacking, and the role of maintenance must be clarified, as well as the positioning of ASCT in the new scenario. A validated definition of hematologic progression would be useful in patient care and in the design of future trials. Further advancements in anti-PC therapy is likely to be based on depth of response, and validation of MS-based detection of monoclonal components and of MRD assessment is warranted to establish optimal goals of therapy. Newer anti-PC approaches, including CAR-T cells and antibody drug conjugates are being considered. Exploration of additional treatment targets, such as interference with LC toxicity by LC stabilizers or doxycycline, which may also target amyloid deposits and is currently undergoing testing in a randomized trial (#NCT03474458), should not be abandoned.
Correspondence
Giovanni Palladini, Amyloidosis Research and Treatment Center, Foundation IRCCS Policlinico San Matteo, Viale Golgi 19, 27100 Pavia, Italy; e-mail: giovanni.palladini@unipv.it.

