

Abstract
The management of Waldenström macroglobulinemia (WM) has evolved tremendously with recent genomic discoveries that correlate with clinical presentation and could help to tailor treatment approaches. The current diagnosis of WM requires clinicopathological criteria, including bone marrow involvement by lymphoplasmacytic lymphoma cells, a serum immunoglobulin M (IgM) monoclonal paraprotein, and presence of the MYD88 L265P mutation. Once the diagnosis is established, the relationship between the patient’s symptoms and WM should be carefully investigated, because therapy should be reserved for symptomatic patients. Bone marrow involvement and serum levels of IgM, albumin, and β2-microglobulin can be used to estimate the time until treatment initiation. The treatment of WM patients should be highly personalized, and the patient’s clinical presentation, comorbidities, genomic profile, and preferences, as well as toxicity of the treatment regimens, should be taken into account. Alkylating agents (bendamustine, cyclophosphamide), proteasome inhibitors (bortezomib, carfilzomib, ixazomib), anti-CD20 monoclonal antibodies (rituximab, ofatumumab), and Bruton tyrosine kinase (BTK) inhibitors (ibrutinib, acalabrutinib, zanubrutinib) are safe and highly effective treatment options in patients with WM. Because novel covalent and noncovalent BTK inhibitors (tirabrutinib, vecabrutinib, LOXO-305, ARQ-531), BCL2 antagonists (venetoclax), and CXCR4-targeting agents (ulocuplumab, mavorixafor) are undergoing clinical development in WM, the future of WM therapy certainly appears bright and hopeful.
Learning Objectives
Describe in detail the criteria for establishing the diagnosis of WM, as well as indications to treat
Review current and upcoming treatment options for patients with symptomatic WM, focusing on the impact of genomic-driven therapies
Clinical case
A 66-year-old asymptomatic man underwent a routine physical examination and was found to have a high serum protein level. Serum protein electrophoresis detected an immunoglobulin M (IgM) κ monoclonal paraprotein. Complete blood count and renal and hepatic function tests were normal. The patient was referred to a hematologist/oncologist for further workup. Serum IgM level was 3500 mg/dL, serum albumin level was 4 g/dL, and serum β2-microglobulin level was 2.5 mg/L. A bone marrow biopsy was performed and showed 40% involvement by κ-restricted lymphocytes and lymphoplasmacytoid cells with positive CD20 and CD38 expression and negative CD5 and CD10 expression, consistent with lymphoplasmacytic lymphoma (LPL). The MYD88 L265P mutation was detected by polymerase chain restriction assay. CXCR4 mutations were not evaluated. Computed tomography (CT) scans of the chest, abdomen, and pelvis showed no evidence of lymphadenopathy or organomegaly. A funduscopic examination did not show evidence of hyperviscosity-related changes.
Initial management
The first step in the management of Waldenström macroglobulinemia (WM) is to properly establish the diagnosis. Based on criteria from the Second International Workshop for Waldenström macroglobulinemia (IWWM), a bone marrow lymphoplasmacytic infiltrate of any level and an IgM monoclonal paraprotein of any size are required for WM diagnosis.1 LPL typically has an intertrabecular pattern of bone marrow infiltration, and the immunophenotype is characterized by positive expression of surface IgM, CD19, CD20, CD22 (dim), CD25, and CD27 and negative expression of CD5, CD10, CD23, and CD103.2 Approximately 5% of patients with LPL will secrete a different protein than IgM and are not considered to have WM. However, the clinical features of non-IgM LPL are similar to WM, although non-IgM LPL patients are less likely to develop neuropathy or hyperviscosity and also have similar outcomes.3 Therefore, the management of non-IgM LPL should follow the guidelines for WM. The MYD88 L265P mutation is detected in >90% of WM patients.4-7 On the other hand, MYD88 mutations are detected in 5% to 10% of patients with chronic lymphocytic leukemia (CLL) or marginal zone lymphoma, and no MYD88 mutations have been detected in multiple myeloma. Non-L265P MYD88 mutations have been described in WM patients, and testing requires sequencing of the entire MYD88 gene.8 In this case, with an elevated serum IgM level, a lymphoplasmacytic infiltrate of the bone marrow, and presence of the MYD88 L265P mutation, the diagnosis of WM is confirmed.
The second step in the management of WM patients is to establish a relationship between the patient’s symptoms, if any, and the underlying disease.9 Asymptomatic or minimally symptomatic WM patients should not be treated. Reasons behind this recommendation include disease incurability, prolonged survival of patients, and toxicity and promotion of resistance associated with therapy. Common indications to treat WM patients include symptomatic anemia, lymphadenopathy, hyperviscosity, or neuropathy.10 Symptomatic cryoglobulinemia, cold agglutinin disease, renal dysfunction, amyloidosis, pleural effusions, and central nervous system involvement are uncommon indications to treat. In our case, the patient is asymptomatic, not anemic, and without evidence of extramedullary disease or hyperviscosity. Therefore, treatment is not indicated. In these situations, the risk of progression to symptomatic disease should be estimated.11 Given the patient’s serum IgM level, percentage of bone marrow involvement, and serum albumin and β2-microglobulin levels, the patient would fall into an intermediate-risk category, with an estimated median time to symptomatic disease ∼5 years. Monitoring without intervention is a reasonable approach. Patients in this setting can be seen every 3 months for clinical evaluations, including symptom reporting, physical examination, and laboratory studies, such as complete blood counts, comprehensive metabolic panel, and serum immunoglobulin levels. Yearly funduscopic examinations are recommended in all WM patients with serum IgM levels ≥ 3000 mg/dL, because the risk of developing symptomatic hyperviscosity appeared to be negligible at lower levels.12
Clinical case (continued)
The patient was clinically evaluated every 3 months and underwent yearly funduscopic examinations. Three years later, the patient presented with recurrent nosebleeds and progressive fatigue affecting his activities. Hemoglobin was 9.2 g/dL, platelets were 115 000 per microliter, and serum IgM level was 5500 mg/dL. There was no evidence of hemolysis or iron, cobalamin, or folate deficiency. Hepatitis B, hepatitis C, and HIV testing was negative. Funduscopic examination revealed engorgement of retinal vessels and scattered retinal microhemorrhages bilaterally. A bone marrow biopsy showed 80% involvement by LPL. MYD88 L265P was detected by polymerase chain reaction, and CXCR4 T318fs (frameshift) was detected by next-generation sequencing assays. CT scans of the chest, abdomen, and pelvis showed generalized lymphadenopathy, with maximum diameter of 3 cm, without hepatosplenomegaly.
Frontline treatment approach
At this time, the patient meets the criteria for treatment initiation, given his symptomatic anemia and evidence of hyperviscosity, according to the guidelines by the Second IWWM.10 Because other causes of anemia and thrombocytopenia have been ruled out, we can assume that the patient’s cytopenias are related to WM. Given the symptoms of hyperviscosity, prompt initiation of plasmapheresis is appropriate to prevent potential thrombotic and/or hemorrhagic complications.13 Plasmapheresis, however, does not constitute definitive treatment of active WM and should be used as a transition toward primary therapy.13 In this setting, screening tests for acquired von Willebrand disease (vWD), such as von Willebrand antigen, ristocetin cofactor, and factor VIII levels, should be performed. Patients with high serum IgM levels and CXCR4 mutations had a higher incidence of acquired vWD,14 which increases the risk of bleeding complications with surgical procedures. The levels of vWD markers typically improve with decreasing serum IgM levels on therapy.
There are several primary therapy options for patients with active symptomatic WM, and the safety and efficacy profiles of selected regimens are shown in Table 1. All patients with WM should be considered for clinical trials, whenever appropriate.15 A suggested treatment algorithm for treatment-naive WM patients is shown in Figure 1. In this case, a treatment regimen associated with a rapid decrease in serum IgM levels would be preferred. Single-agent rituximab is less effective in WM patients with serum IgM levels ≥ 4000 mg/dL, and the median time to response ranges between 3 and 6 months.16 Also, 40% to 50% of WM patients exposed to single-agent rituximab can experience an IgM flare, which can induce rapid increases in serum IgM ranging from 25% to 300% and could worsen hyperviscosity symptoms.17 In this setting, alkylating agents or proteasome inhibitors in combination with rituximab, as well as ibrutinib with and without rituximab, are reasonable options.
Selected treatment regimens for patients with WM
| Study | Agent | N (TN/RR) | ORR, % | MRR, % | VGPR, % | PFS | Adverse events |
|---|---|---|---|---|---|---|---|
| Dimopoulos et al44 | Cyclophosphamide, D, R | 72 (72/0) | 83 | 74 | 7 | Median: 35 mo | Cytopenias, infections, myeloid neoplasms |
| Rummel et al45 | Bendamustine, R | 19 (19/0) | NR | NR | NR | Median: 69.5 mo | |
| R-CHOP | 22 (22/0) | NR | NR | NR | Median: 28 mo | ||
| Rummel et al27 | Bendamustine, R | 257 (257/0) | 92 | 88 | 4 | Median: 65 mo | |
| Treon et al22 | Bortezomib (twice weekly), D, R | 23 (23/0) | 96 | 83 | 22 | Median: 66 mo | Neuropathy, neutropenia, infections |
| Dimopoulos et al46 | Bortezomib (weekly), D, R | 59 (59/0) | 85 | 58 | 10 | Median: 42 mo | |
| Treon et al47 | Carfilzomib, D, R | 31 (31/0) | 87 | 68 | 35 | Median: 44 mo | Hyperglycemia, hyperlipasemia |
| Castillo et al48 | Ixazomib, D, R | 26 (26/0) | 96 | 77 | 15 | Median: NR at 22 mo | Infections, hyperglycemia |
| Treon et al28,29 | Ibrutinib | 63 (0/63) | 91 | 81 | 16 | 5 y: 54% | Cytopenias, bleeding, arrhythmias, hypertension |
| Treon et al36 | Ibrutinib | 30 (30/0) | 100 | 83 | 20 | 18 mo: 92% | |
| Dimopoulos et al34 | Ibrutinib, R | 75 (34/41) | 93 | 73 | 26 | 30 mo: 82% | |
| Owen et al37 | Acalabrutinib | 106 (14/92) | 93 | 78 | 8 (IWWM6) 29 (IWWM3) | 24 mo: 90% (TN); 82% (RR) |
| Study | Agent | N (TN/RR) | ORR, % | MRR, % | VGPR, % | PFS | Adverse events |
|---|---|---|---|---|---|---|---|
| Dimopoulos et al44 | Cyclophosphamide, D, R | 72 (72/0) | 83 | 74 | 7 | Median: 35 mo | Cytopenias, infections, myeloid neoplasms |
| Rummel et al45 | Bendamustine, R | 19 (19/0) | NR | NR | NR | Median: 69.5 mo | |
| R-CHOP | 22 (22/0) | NR | NR | NR | Median: 28 mo | ||
| Rummel et al27 | Bendamustine, R | 257 (257/0) | 92 | 88 | 4 | Median: 65 mo | |
| Treon et al22 | Bortezomib (twice weekly), D, R | 23 (23/0) | 96 | 83 | 22 | Median: 66 mo | Neuropathy, neutropenia, infections |
| Dimopoulos et al46 | Bortezomib (weekly), D, R | 59 (59/0) | 85 | 58 | 10 | Median: 42 mo | |
| Treon et al47 | Carfilzomib, D, R | 31 (31/0) | 87 | 68 | 35 | Median: 44 mo | Hyperglycemia, hyperlipasemia |
| Castillo et al48 | Ixazomib, D, R | 26 (26/0) | 96 | 77 | 15 | Median: NR at 22 mo | Infections, hyperglycemia |
| Treon et al28,29 | Ibrutinib | 63 (0/63) | 91 | 81 | 16 | 5 y: 54% | Cytopenias, bleeding, arrhythmias, hypertension |
| Treon et al36 | Ibrutinib | 30 (30/0) | 100 | 83 | 20 | 18 mo: 92% | |
| Dimopoulos et al34 | Ibrutinib, R | 75 (34/41) | 93 | 73 | 26 | 30 mo: 82% | |
| Owen et al37 | Acalabrutinib | 106 (14/92) | 93 | 78 | 8 (IWWM6) 29 (IWWM3) | 24 mo: 90% (TN); 82% (RR) |
D, dexamethasone; MRR, major response rate; NR, not reported; R, rituximab; R-CHOP, rituximab, cyclophosphamide, doxorubucin, vincristine, and prednisone; RR, relapsed/refractory; TN, treatment naive.
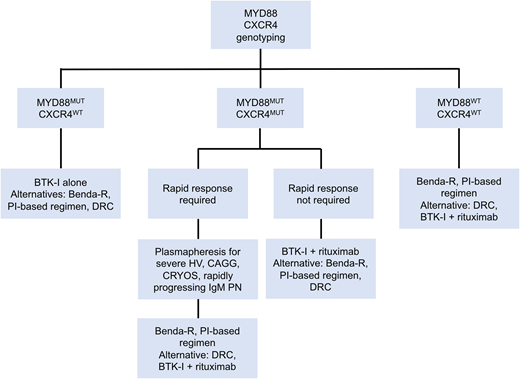
Genomic-based treatment algorithm for symptomatic treatment-naive patients with WM. Benda-R, bendamustine and rituximab; BTK-I, Bruton tyrosine kinase inhibitor; CAGG, cold agglutinin disease; CRYOS, cryoglobulins; DRC, dexamethasone, rituximab, cyclophosphamide; HV, hyperviscosity; PI, proteasome inhibitor; PN, progressive neuropathy. Adapted with permission from Treon et al.49
Genomic-based treatment algorithm for symptomatic treatment-naive patients with WM. Benda-R, bendamustine and rituximab; BTK-I, Bruton tyrosine kinase inhibitor; CAGG, cold agglutinin disease; CRYOS, cryoglobulins; DRC, dexamethasone, rituximab, cyclophosphamide; HV, hyperviscosity; PI, proteasome inhibitor; PN, progressive neuropathy. Adapted with permission from Treon et al.49
A careful and thorough discussion between practitioners and patients should take place on the positive and negative aspects of each treatment option. All of the regimens mentioned above are associated with high overall and major response rates. Therapy selection in WM patients should be personalized, taking into account the patient’s symptoms, comorbidities, genomic profile, preferences, and insurance coverage, as well as the safety profile of the regimen. MYD88 wild-type and CXCR4 mutated status have been associated with lower efficacy rates with ibrutinib monotherapy.8,18 CXCR4 mutations do not seem to impact progression-free survival (PFS) on alkylator-based or proteasome inhibitor–based regimens.19,20 Alkylators are associated with a 1% to 2% risk for myeloid neoplasms, bortezomib is associated with a 20% to 25% risk for peripheral neuropathy, and ibrutinib is associated with a 5% to 10% risk for atrial fibrillation.21-23 The administration of these agents also differs; bendamustine is administered IV and bortezomib is given subcutaneously and both are of finite duration, whereas ibrutinib is an oral agent of indefinite duration.
The role of maintenance rituximab therapy after induction chemoimmunotherapy in WM patients continues to evolve. Several retrospective studies have suggested a deepening of response, as well as PFS and overall survival benefits in WM patients treated with maintenance rituximab vs observation after rituximab-containing regimens.24-26 However, preliminary data from the MAINTAIN study, presented at the 2019 American Society of Hematology Annual Meeting, did not find any PFS or overall survival benefit from maintenance rituximab vs observation after attaining a partial response or better to bendamustine and rituximab.27 It is important to note that patients who attained a minor response after induction were not randomized and that patients older than 65 years or with high-risk disease, based on the International Prognostic Scoring System for WM, seemed to have derived survival benefit from maintenance therapy.
Clinical case (continued)
The patient went on to receive 6 cycles of bendamustine and rituximab. At the end of therapy, the patient’s blood counts normalized, his lymphadenopathy resolved, and his serum IgM level was 1400 mg/dL, consistent with a partial response. His symptoms also resolved, and the patient was monitored every 3 months. Three years later, the patient presented with progressive fatigue and symptomatic anemia. His hemoglobin level was 9.7 g/dL, his platelet count was 110 000 per microliter, and his serum IgM level was 3400 mg/dL. Funduscopic examination did not show changes associated with hyperviscosity. CT scans did not show any evidence of lymphadenopathy or organomegaly. A bone marrow aspiration and biopsy showed 80% involvement by LPL, without evidence of dysplasia. MYD88 L265P and a frameshift CXCR4 mutation were detected.
Treatment options in the relapsed setting
Current treatment options for patients with previously treated WM are highly effective. Selected regimens in this setting are shown in Table 1. A suggested treatment algorithm for previously treated WM patients is shown in Figure 2. As with primary therapy, a personalized approach should be followed when selecting treatments for patients with relapsed WM. Given prior exposure to alkylating agents, Bruton tyrosine kinase (BTK) inhibitors are reasonable in this setting, because they have been associated with response rates well over 90% and median PFS in excess of 5 years.28,29 In the pivotal phase 2 study of 63 relapsed WM patients, ibrutinib monotherapy, at a dose of 420 mg by mouth every day, was associated with high overall response rate (ORR), major response, and very good partial response (VGPR) rate, with an estimated 2-year PFS of 69%. These results paved the way for the US Food and Drug Administration approval of ibrutinib in symptomatic WM patients in April of 2015. Long-term data from this study were presented at the 2019 Lugano Conference29 and showed deepening of major response and VGPR, with a 5-year PFS rate of 54%. Patients with MYD88 mutation and without CXCR4 mutations had higher ORR, major response rate, and 5-year PFS to ibrutinib monotherapy than patients with MYD88 and CXCR4 mutations. Although CXCR4 mutations adversely impact depth and duration of response to ibrutinib, patients with frameshift CXCR4 mutations (rather than non-sense mutations) seem to derive similar benefits from ibrutinib therapy as do patients without CXCR4 mutations.18
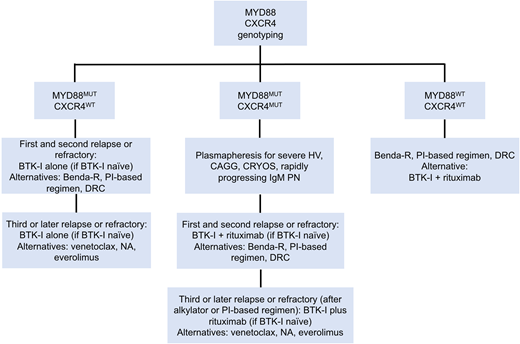
Genomic-based treatment algorithm for symptomatic, previously treated, or refractory patients with WM. Benda-R, bendamustine and rituximab; BTK-I, BTK inhibitor; CAGG, cold agglutinin disease; CRYOS, cryoglobulins; DRC, dexamethasone, rituximab, cyclophosphamide; HV, hyperviscosity; NA, nucleoside analogs; PI, proteasome inhibitor; PN, progressive neuropathy. Adapted with permission from Treon et al.49
Genomic-based treatment algorithm for symptomatic, previously treated, or refractory patients with WM. Benda-R, bendamustine and rituximab; BTK-I, BTK inhibitor; CAGG, cold agglutinin disease; CRYOS, cryoglobulins; DRC, dexamethasone, rituximab, cyclophosphamide; HV, hyperviscosity; NA, nucleoside analogs; PI, proteasome inhibitor; PN, progressive neuropathy. Adapted with permission from Treon et al.49
However, one must be aware of specific side effects associated with ibrutinib therapy. Early side effects include rash, diarrhea, abdominal bloating, and nausea, which improve and resolve within a few weeks on therapy in most patients. Long-term side effects include bleeding, arrhythmia, and withdrawal symptoms. Ibrutinib affects platelet aggregation and adhesion,30 increasing the risk of bleeding with surgical procedures, and it should be held temporarily for a few days before and after each procedure to minimize bleeding risk. Ibrutinib has also been associated with an increased risk for arrhythmia, especially atrial fibrillation.31 Bετα-blockers, anticoagulants, antiarrhythmics, and/or cardiac ablation can be used, if necessary, under the care of a cardiologist with experience with this complication. An algorithm for the management of ibrutinib-related atrial fibrillation has been published.32 About 20% of WM patients who discontinue ibrutinib temporarily might experience withdrawal symptoms, such as fever, night sweats, and fatigue, which could be managed with low doses of steroids during the hold.33 An increase in serum IgM levels can also be seen during holds and should not be considered disease progression, because serum IgM levels decrease promptly after restarting ibrutinib.
A multicenter randomized phase 3 study (INNOVATE) evaluated the combination of ibrutinib and rituximab vs placebo and rituximab in 150 patients with WM.34 The combination of ibrutinib and rituximab was associated with a higher ORR (92% vs 47%) and major response rate (72% vs 32%), as well as higher 30-month PFS (82% vs 28%), compared with placebo and rituximab. There were higher rates of atrial fibrillation, hypertension, and serious respiratory infections and lower rates of infusion-related reactions and IgM flare in patients who received ibrutinib and rituximab compared with placebo and rituximab. Based on these results, the US Food and Drug Administration approved the combination of ibrutinib and rituximab for symptomatic WM in August of 2018. The results of that study do not address whether the combination of ibrutinib and rituximab is superior to ibrutinib monotherapy in WM patients, and it is unlikely that a study addressing that question in WM patients will ever be done. In a randomized study of patients with CLL, the combination of ibrutinib and rituximab was not associated with superior response rates or longer PFS compared with ibrutinib alone,35 making the addition of rituximab to ibrutinib of unclear long-term benefit in CLL. On the other hand, the combination of ibrutinib and rituximab induced major responses in 54% of the 16 WM patients without MYD88 mutation, whereas the major response rate to ibrutinib alone was 0% in 5 WM patients without MYD88 mutation.8 In patients with CXCR4 mutations, the median time to major response with the combination of ibrutinib and rituximab was 3 months, whereas prior studies had reported a median time to response of 6 to 7 months with ibrutinib monotherapy.29,36 It is unclear whether the addition of rituximab to ibrutinib would benefit all patients. The combination of ibrutinib and rituximab can be considered in WM patients with CXCR4 mutations or in MYD88 wild-type patients.
Clinical case (continued)
The patient was started on ibrutinib, 420 mg by mouth every day. Within 3 months of therapy, the patient’s hemoglobin normalized, and his serum IgM level decreased to 320 mg/dL, consistent with a VGPR to therapy. The patient remains on ibrutinib monotherapy.
Future treatment options
Despite the depth of response attained by the patient within the first 3 months of therapy, one could expect progression of disease at some point in the future. Therefore, additional research is needed to identify novel treatment options. Selected ongoing clinical trials are shown in Table 2.
Selected ongoing clinical trials with novel agents in patients with WM
| ClinicalTrials.Gov ID | Agents | Mechanism of action | Phase |
|---|---|---|---|
| NCT04263480 | Ibrutinib Carfilzomib | BTK inhibitor Proteasome inhibitor | 3 |
| Ibrutinib | BTK inhibitor | ||
| NCT04061512 | Ibrutinib Rituximab | BTK inhibitor Anti-CD20 mAb | 2/3 |
| Cyclophosphamide Rituximab Dexamethasone | Alkylating agent Anti-CD20 mAb Steroid | ||
| NCT03506373 | Ibrutinib Ixazomib | BTK inhibitor Proteasome inhibitor | 2 |
| NCT03620903 | Ibrutinib Bortezomib Rituximab | BTK inhibitor Proteasome inhibitor Anti-CD20 mAb | 2 |
| NCT04273139 | Ibrutinib Venetoclax | BTK inhibitor BCL2 antagonist | 2 |
| NCT03679624 | Ibrutinib Daratumumab | BTK inhibitor Anti-CD38 mAb | 2 |
| NCT03187262 | Daratumumab | Anti-CD38 mAb | 2 |
| NCT03630042 | Pembrolizumab Rituximab | Anti-PD1 mAb Anti-CD20 mAb | 2 |
| NCT02962401 | Idelalisib Obinutuzumab | PI3K inhibitor Anti-CD20 mAb | 2 |
| NCT03364231 | Umbralisib | PI3K inhibitor | 2 |
| NCT03225716 | Ibrutinib Ulocuplumab | BTK inhibitor Anti-CXCR4 mAb | 1/2 |
| NCT02457559 | Tirabrutinib | BTK inhibitor | 1/2 |
| NCT03037645 | Vecabrutinib | BTK inhibitor | 1/2 |
| NCT03162536 | ARQ-351 | BTK inhibitor | 1/2 |
| NCT03740529 | LOXO-305 | BTK inhibitor | 1/2 |
| NCT04274738 | Ibrutinib Mavorixafor | BTK inhibitor CXCR4 antagonist | 1 |
| NCT04115059 | Dasatinib | HCK inhibitor | Pilot |
| ClinicalTrials.Gov ID | Agents | Mechanism of action | Phase |
|---|---|---|---|
| NCT04263480 | Ibrutinib Carfilzomib | BTK inhibitor Proteasome inhibitor | 3 |
| Ibrutinib | BTK inhibitor | ||
| NCT04061512 | Ibrutinib Rituximab | BTK inhibitor Anti-CD20 mAb | 2/3 |
| Cyclophosphamide Rituximab Dexamethasone | Alkylating agent Anti-CD20 mAb Steroid | ||
| NCT03506373 | Ibrutinib Ixazomib | BTK inhibitor Proteasome inhibitor | 2 |
| NCT03620903 | Ibrutinib Bortezomib Rituximab | BTK inhibitor Proteasome inhibitor Anti-CD20 mAb | 2 |
| NCT04273139 | Ibrutinib Venetoclax | BTK inhibitor BCL2 antagonist | 2 |
| NCT03679624 | Ibrutinib Daratumumab | BTK inhibitor Anti-CD38 mAb | 2 |
| NCT03187262 | Daratumumab | Anti-CD38 mAb | 2 |
| NCT03630042 | Pembrolizumab Rituximab | Anti-PD1 mAb Anti-CD20 mAb | 2 |
| NCT02962401 | Idelalisib Obinutuzumab | PI3K inhibitor Anti-CD20 mAb | 2 |
| NCT03364231 | Umbralisib | PI3K inhibitor | 2 |
| NCT03225716 | Ibrutinib Ulocuplumab | BTK inhibitor Anti-CXCR4 mAb | 1/2 |
| NCT02457559 | Tirabrutinib | BTK inhibitor | 1/2 |
| NCT03037645 | Vecabrutinib | BTK inhibitor | 1/2 |
| NCT03162536 | ARQ-351 | BTK inhibitor | 1/2 |
| NCT03740529 | LOXO-305 | BTK inhibitor | 1/2 |
| NCT04274738 | Ibrutinib Mavorixafor | BTK inhibitor CXCR4 antagonist | 1 |
| NCT04115059 | Dasatinib | HCK inhibitor | Pilot |
HCK, hematopoietic cell kinase; mAb, monoclonal antibody; PD1, programmed cell death protein 1; PI3K, phosphatidylinositol-3 kinase.
Ibrutinib is being evaluated in combination with chemoimmunotherapy, proteasome inhibitors, BCL2 inhibitors, and anti-CD38 antibodies. Acalabrutinib, zanubrutinib, and tirabrutinib are covalent BTK inhibitors also being studied in WM patients. A large multicenter phase 2 study evaluated acalabrutinib in 106 WM patients and reported an ORR of 93%, major response of 80%, and 2-year PFS rate of 80% to 90%.37 Most common grade ≥3 adverse events included neutropenia and lower respiratory tract infections. The rate of atrial fibrillation was 5%. A phase 1/2 prospective study evaluated zanubrutinib in 77 WM patients.38 Zanubrutinib was associated with an ORR of 92%, major response rate of 82%, VGPR rate of 41%, and 24-month PFS rate of 82%. Adverse events of bruising/bleeding and atrial fibrillation (5%) were observed. A randomized phase 3 study evaluating zanubrutinib (Arm A) vs ibrutinib (Arm B) in symptomatic WM patients (ASPEN) has completed accrual.39 At 19 months of follow-up, VGPR rates for zanubrutinib and ibrutinib were 28% and 19%, respectively, and 12-month PFS rates were 90% and 87%, respectively. There were lower rates of atrial fibrillation, diarrhea, and bleeding, but higher rates of neutropenia, with zanubrutinib. Preliminary results of ASPEN Arm C showed that zanubrutinib induced responses in patients without MYD88 mutations, with an ORR of 77%, major response of 54%, and VGPR rate of 15%.40 Tirabrutinib was evaluated in 27 patients with WM.41 ORR was 94% and 100%, and major response rates were 78% and 89% in treatment-naive and previously treated patients, respectively. Rash was reported in 41% of patients. The acquisition of BTK mutations has been associated with resistance to covalent BTK inhibitors in patients with WM.42 Second-generation noncovalent BTK inhibitors (eg, vecabrutinib, LOXO-305, ARQ-531) are being investigated in WM patients. A multicenter prospective phase 2 clinical trial evaluating a 2-year course of venetoclax in 30 previously treated WM patients has completed accrual.43 Preliminary results showed ORR, major response rate, and VGPR rate of 90%, 83%, and 20%, respectively, and 18-month PFS rate of 82%. Grade ≥3 adverse events included neutropenia, anemia, and diarrhea. Studies evaluating CXCR4-targeting agents, such as ulocuplumab (monoclonal antibody) and mavorixafor (small molecule), are ongoing.
In conclusion, there have been a series of advances in the diagnosis and management of WM in recent years. Rational genomic-driven treatment options are increasing in number, and it is hoped that they will translate into deeper and more durable responses, as well as lower toxicity rates.
Acknowledgments
The authors thank the laboratory and clinical staff at the Bing Center for Waldenström Macroglobulinemia for their tireless efforts and support, and the patients of the Waldenström Macroglobulinemia Clinic, who are the reason why we do what we do.
Correspondence
Jorge J. Castillo, Dana-Farber Cancer Institute, 450 Brookline Ave, Mayer 221, Boston, MA 02215; e-mail. jorgej_castillo@dfci.harvard.edu.
