Abstract
Immunotherapy is now a well-established modality in the treatment of cancer. Although several platforms to redirect the immune response exist, the use of genetically modified T cells has garnered particular attention in recent years. This is due, in large part, to their success in the treatment of B-cell malignancies. Adoptively transferred T cells have also demonstrated efficacy in the treatment of systemic viral infections that occur following hematopoietic cell transplantation prior to immune reconstitution. Here we discuss the techniques that enable redirection of T lymphocytes to treat cancer or infection and the current indications for these therapies.
Learning Objectives
Describe the methods that enable production of redirected T cells
Review the current indications for approved T-cell products
CLINICAL CASE
A 56-year-old man seeks treatment from his primary care physician for 2 months of progressive fatigue and night sweats. Physical examination reveals axillary lymphadenopathy. Laboratory studies reveal only an elevated lactate dehydrogenase but are otherwise unremarkable. Positron emission tomography (PET) reveals several sites of nodal fluorodeoxyglucose- uptake. Lymph node biopsy specimen ultimately reveals CD19+ CD10+ BCL6+ mature B cells with histopathologic and cytogenetic features consistent with germinal center-type large B-cell lymphoma. His Eastern Cooperative Oncology Group (ECOG) performance status is assessed as 1, and he initiates chemoimmunotherapy with combination rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP). After 6 cycles, he has evidence of disease progression and undergoes salvage chemotherapy with rituximab, ifosfamide, carboplatin, and etoposide (R-ICE). After 2 cycles, PET reveals persistent progressive disease, demonstrating chemotherapy resistance.
T cells as therapeutic agents
Several cellular attributes make T cells extremely effective at their native function of controlling infection. First, T cells are endowed with the ability to differentiate “self” from “non-self” antigens, reflecting exquisite specificity. This determination is made by the surface membrane antigen receptor called the T-cell receptor (TCR). In a manner similar to antibody production, the large genes encoding the TCR undergo somatic recombination, a process in which the multicomponent TCR genes are edited to produce smaller genes. This rearrangement results in the production of two recombined genes from more than 1018 potential rearrangements, endowing each individual T-cell clone with an essentially unique TCR. In addition to this antigen specificity, T cells are potent killers of target cells, ensuring efficient viral eradication. Upon antigen recognition, T cells undergo rapid proliferation to generate a large cytotoxic immune response. Finally, antigen-specific T cells are able to differentiate into long-lived memory cells, capable of mounting memory responses decades after the initial infectious insult. Together, these features not only make T cells highly evolved and efficient controllers of infection but also make them very attractive as anticancer agents.
Although T cells have great potential to serve as antileukemic agents by recognition of host leukemia as “non-self” (this is the primary therapeutic effect of allogeneic hematopoietic cell transplantation [HCT]), they also recognize healthy host tissues as foreign, resulting in the syndrome of graft-versus-host disease. Extrication of these 2 processes, graft-versus-leukemia and graft-versus-host, has given rise to the field of T-cell engineering for the purpose of redirecting this potent immune response toward cancer or uncontrolled viral infection.
Redirecting the T-cell response
The processes of engineering T cells for use as anticancer or antiviral therapeutics are fundamentally distinct and will be addressed separately.
Cancer
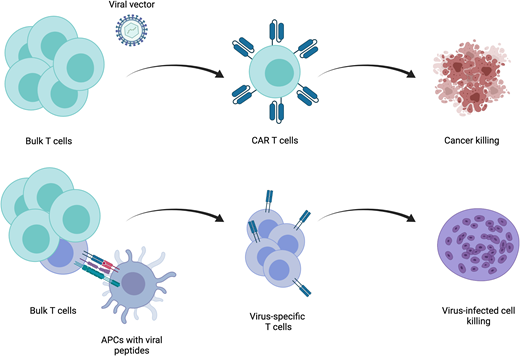
Redirection of the T-cell response is dependent on the type of antigen they are intended to target. Antigens fall into 2 broad categories—intracellular and cell surface. For a T cell to recognize proteins that are intracellular, these proteins must be processed and presented by major histocompatibility complex (MHC) proteins for recognition by TCRs. Introduction of a transgenic TCR that has been designed, or more often identified among naturally occurring TCRs, to be specific for a cancer-associated antigen allows for recognition of intracellular cancer antigens. This process, which reconstructs all components of the native TCR complex to activate the T-cell response, has thus far been met with limited success. Identification of immunogenic antigens and TCR cognates is difficult. Furthermore, identification of cancer-specific antigens is unusual, and cancer cells often downregulate surface MHC. Although several studies have evaluated the efficacy of these agents,1 disease control has been modest. For these reasons, an approach using synthetic antigen receptors that are not dependent on MHC has been developed. The most well developed of these are chimeric antigen receptors (CARs). These hybrid receptors contain an antigen recognition domain derived from a monoclonal antibody. This strategy allows for MHC-independent antigen recognition while preserving much of the native TCR-driven activation machinery. The downside, of course, is loss of the ability to target intracellular antigens. This extracellular antigen-binding domain is linked to intracellular T-cell activating domains. Currently, only 2 signaling domain structures are used in the several CAR T-cell products approved for use by the US Food and Drug Administration (FDA). Both contain the CD3ζ signaling domain, which recapitulates the classical “signal 1” of T-cell stimulation through the TCR. These 2 structures differ in the composition of their costimulatory domains, one employing CD28 and the other 41BB. Correlative data from clinical trials demonstrate that CD28-based CARs tend to undergo greater and more rapid expansion after delivery but do not persist for long periods. Alternatively, 41BB-based CARs do not expand as quickly or to the same degree but can persist for years. Both CAR- and TCR-based approaches rely on genetic engineering to deliver either of these transgenic receptors to T cells. Significant advances in gene therapy using viral vectors have enabled efficient delivery of genes encoding these receptors to T cells.
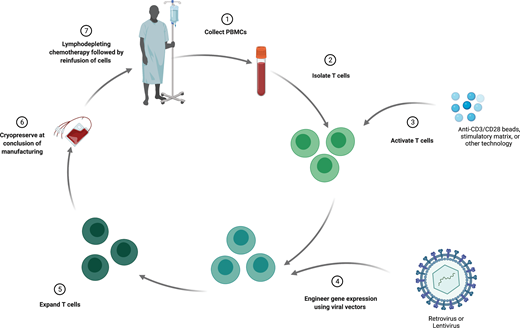
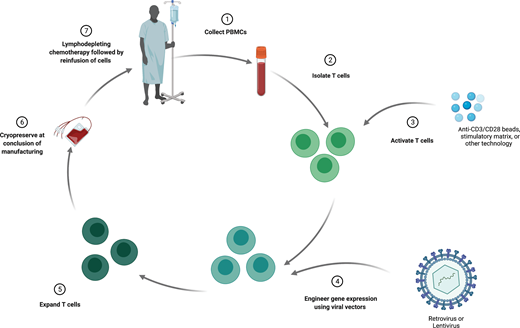
The process of manufacturing autologous T cells with synthetic antigen receptors (Figure 1) begins with an initial leukapheresis to isolate peripheral blood mononuclear cells (PBMCs). This PBMC product undergoes processing to purify T cells, which are then cultured with a stimulatory agent, often magnetic beads containing stimulatory antibodies directed at CD3 and CD28. This stimulation induces T-cell activation, DNA replication, and proliferation. Following stimulation, gene therapy vectors (most often lentivirus or retrovirus) are added to the T-cell cultures, which mediate transfer of the target transgene. These engineered cells are then grown in culture for several days and finally are cryopreserved for eventual infusion. All of these steps, whether at an academic center for treatment on clinical trials or at a commercial production facility, are performed under Good Manufacturing Practice guidelines. At several time points during this manufacturing process, quality control measures are performed to ensure Good Manufacturing Practice standards. Prior to delivery of the T-cell product, patients receive lymphodepleting chemotherapy. Similar to the concept in allogeneic HCT, this lymphodepletion facilitates successful engraftment of the engineered product. This step can also further aid the therapeutic response by reducing disease burden, as CAR T cells are still most often used for lymphoid malignancies.
Infection
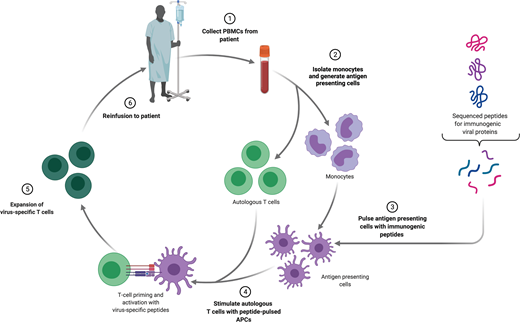
The primary indication for adoptive T-cell immunotherapy in the treatment of infectious diseases is clinically significant viral infection following HCT. Over the past 2 decades, strategies to treat viruses that are responsible for significant morbidity and mortality, such as cytomegalovirus,2 Epstein-Barr virus (EBV),3 adenovirus,4,5 human herpes virus 6,6 BK virus,7 varicella zoster virus,8 or broad-spectrum antiviral treatments,9 have been developed. Most studies have been performed using autologous cells, derived from either the patient themselves in the setting of autologous transplantation or the donor in the setting of allogeneic transplantation. The fundamental principle underlying the generation of virus-specific T cells (VSTs) in both settings is to expand a preexisting population of antigen-specific memory T cells. This requires that the patient or donor has previously been exposed to that virus and an appropriate method to present stimulatory antigens to VSTs.
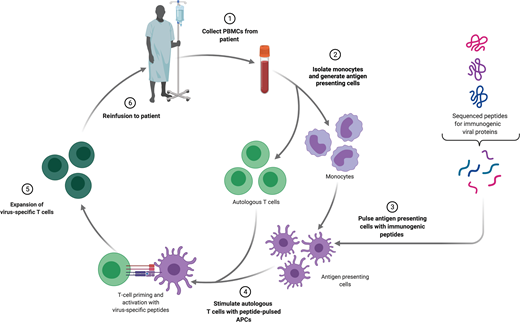
Several strategies exist to isolate and expand VSTs from a bulk T-cell population ex vivo and differ based on the type of antigen and method of antigen presentation (Figure 2). The simplest of these is to deliver a whole viral lysate to antigen presenting cells (APCs), such as dendritic cells, and coculture these with T cells. This approach has limited clinical utility, because there is an underlying risk of transfer of a live infectious particle. An alternative is delivering purified or recombinant viral proteins or plasmid DNA encoding viral proteins to APCs. The most widely used approach employs the use of pools of peptide delivered to APCs.10 After selection of the stimulation strategy, VSTs are most often expanded in repeat stimulation cultures. This process results in the production of a large number of polyclonal T cells, but manufacturing time is often several weeks to even months long. An alternative approach is direct isolation of existing VSTs from peripheral blood. Antigen-specific T cells will bind peptide-HLA multimers, and these multimers can be isolated along with bound T cells. This approach can feasibly only be applied in settings of high VST frequency and as such has limited utility.
Clinical products for the treatment of cancer
The majority of clinical experience using engineering T cells is in the treatment of B-cell malignancies. Currently, 5 CAR T-cell products are approved for commercial use by the FDA; 4 of these target the B-cell antigen CD19, and 1 targets the plasma cell antigen B-cell maturation antigen (BCMA). Although these products differ in several ways, the primary variance is derived from the structure of the receptor's costimulatory domains. In addition to these FDA-approved products, several others have been evaluated in clinical trials. Key findings from these studies are summarized in Table 1.
Acute lymphoblastic leukemia
Non-Hodgkin lymphoma
Four products are approved for the treatment of non-Hodgkin lymphoma in adults. Tisagenlecleucel is indicated for patients with diffuse large B-cell lymphoma (DLBCL), high-grade B-cell lymphoma, or DLBCL arising from follicular lymphoma that is relapsed or refractory after 2 or more lines of therapy.13 Axicabtagene ciloleucel is also directed at CD19 but contains a costimulatory domain derived from CD28.14,15 It is approved for adults with DLBCL, primary mediastinal large B-cell lymphoma, high-grade B-cell lymphoma, and DLBCL arising from follicular lymphoma that is relapsed or refractory after 2 or more lines of therapy. Lisocabtagene maraleucel also contains the 41BB costimulatory domain but is delivered as 2 individual products: one composed of CD4+ and the other of CD8+ T cells.16 It is indicated for all of the lymphoma subtypes mentioned above, as well as grade 3B follicular lymphoma and DLBCL arising from nonfollicular indolent histologies. Finally, brexucabtagene autoleucel is structurally identical to axicabtagene ciloleucel but undergoes an additional B-cell depletion step during product manufacturing and is approved for relapsed or refractory mantle cell lymphoma.17
Multiple myeloma
Idecabtagene vicleucel was recently approved for adult patients with relapsed or refractory multiple myeloma after 4 or more lines of therapy, including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 monoclonal antibody.18 Unlike the other approved therapies, it targets the BCMA on plasma cells. Like tisagenlecleucel and listocabtagene maraleucel, it contains the 41BB costimulatory domain.
Patient selection
Given the availability of CAR T-cell products, particularly in the management of relapsed or refractory non-Hodgkin lymphoma, a key clinical consideration is when to use this therapy and which therapy to use. At many academic centers, CAR T-cell therapy is commonly considered for patients with progressive lymphoma after second-line chemotherapy or in the setting of relapse after autologous transplantation. Decision making about using CAR T cells vs autologous transplantation, specifically in the setting of failed first-line therapy, is an active topic of discussion and will be assisted by the results of current ongoing randomized clinical trials (NCT03391466, NCT03570892, NCT03575351).
Incidence and management of toxicities is also a relevant consideration. The most common toxicity occurring following CAR T-cell therapy is cytokine release syndrome (CRS), a systemic hyperinflammatory syndrome driven by supraphysiologic production of interleukin 6 (IL-6) bystander myeloid-lineage cells in response to robust T-cell activation.19,20 The use of IL-6 and IL-6 receptor inhibitors, such as tocilizumab, has significantly improved outcomes for patients. Another highly morbid toxicity is immune effector cell-associated neurotoxicity syndrome (ICANS),21 a poorly understood neuropsychiatric syndrome that is commonly managed with corticosteroids and antiepileptics.22 In rare cases, ICANS can progress and lead to cerebral edema, a much more serious condition that is primarily managed with high-dose steroids. Intriguingly, both toxicities appear to be more severe with CD28-based CAR T cells, and for this reason, many clinicians will elect to use 41BB-based therapy, if available, for patients who are more frail. Clinical trials of CAR T products only enrolled patients with high-performance statuses (ECOG 0-1). However, real-world data suggest that patients who are not as fit can be successfully treated with CAR therapy.23 Indeed, given the management options now available for CRS and ICANS, most patients are considered eligible for this therapy. Other toxicities include prolonged pancytopenia and a durable loss of B cells with associated hypogammaglobulinemia.
Finally, given the autologous nature of this therapy, product manufacturing is also worth considering. Several studies have demonstrated that both previous exposure to chemotherapy and the immunosuppressive environment encumbered by active malignancy can significantly impair the effective expansion of collected T cells.24,25 Several centers have explored “early banking” procedures, in which high-risk patients undergo apheresis and storage of T cells prior to initiating first-line therapy to ensure availability of “fit” T cells should they be needed. This approach remains investigational and should only be considered in the context of a clinical trial.
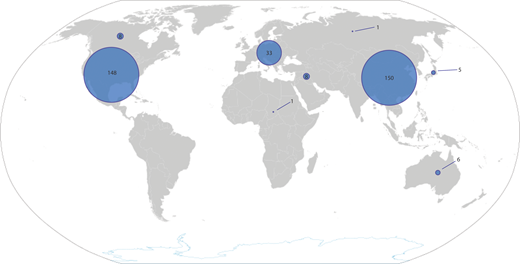
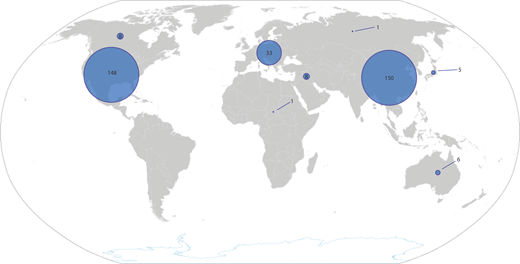
The availability of CAR T-cell products has increased significantly in the past 5 years. For children with ALL, therapy is almost always delivered in an academic setting, which also facilitates availability of tisagenlecleucel. At the time of writing, there were 324 actively recruiting clinical trials of CAR T-cell products. Most of these are in the United States (148) and China (150), reflecting the disparity in access to this platform that exists across the globe (Figure 3).
Clinical products for the treatment of infection
Currently, there are no FDA-approved products available for the treatment of viral infection using T cells. Three phase 3 studies are currently ongoing (NCT04832607, NCT04390113, NCT03394365) evaluating the efficacy of VSTs. One (NCT03394365) is focused on the efficacy of a VST product targeting EBV antigens in the management of EBV-associated posttransplant lymphoproliferative disorder,26 as well chronic active EBV and EBV-driven hemophagocytic lymphohistiocytosis (HLH). Greater than 100 clinical trials at all phases are currently enrolling across the globe. An exciting prospect is the development of allogeneic VSTs that can serve as an “off-the-shelf” treatment, eliminating the time delay between disease onset and treatment.27
CLINICAL CASE (continued)
After discussing the distinctions between autologous transplantation and CAR T cells, the patient elects to proceed with CAR T cells. He undergoes apheresis for planned therapy with tisagenlecleucel, with successful product manufacturing. He receives conditioning chemotherapy with fludarabine and cyclophosphamide, followed by CAR T-cell infusion. He experiences chills and rigors on day +3, classified as grade 1 CRS. These resolve spontaneously on day +5. After count recovery on day +11, he is discharged from the hospital without incurring any further toxicity. Thirty days after treatment, PET/computed tomography reveals a partial response. PET/computed tomography at 3 months reveals complete remission.
Challenges and next-generation T cells for cancer
Although great progress has been made in the use of engineered T cells, particularly with regard to the treatment of hematologic cancers, the reality is that most patients treated with CD19-directed CAR T cells will not achieve durable remissions. The 2 features that have been identified as critical barriers to the broader success of this therapy are (1) initial failure that leads to nonresponse and (2) acquired failure that leads to disease relapse. The former is more common in lymphoma, whereas the latter is more common in ALL. Several studies have identified T cell–intrinsic features that are associated with initial failure, such as expression of dysfunction-associated genes, terminal differentiation, and impaired T-cell persistence.25,28-30 New strategies to enhance T-cell fitness using innovative manufacturing processes have generated great excitement and open avenues to the manufacturing of T cells with enhanced function.31,32
Several mechanisms have been described that lead to disease relapse, most thoroughly the phenomenon of antigen escape. In approximately 40% of patients with ALL who relapse, the recurrent leukemia lacks detectable surface CD19, enabling disease outgrowth.33,34 To overcome antigen escape, several groups have employed dual-antigen targeting to more thoroughly capture malignant clones and reduce the likelihood of antigen loss as a mechanism of relapse (NCT03241940, NCT03330691, NCT03448393).
Conflict-of-interest disclosure
Nathan Singh holds several patents related to CAR and other engineered cell therapies.
Off-label drug use
Nathan Singh: nothing to disclose.