Abstract
Hereditary hemorrhagic telangiectasia (HHT), the second most common inherited bleeding disorder, is associated with the development of malformed blood vessels. Abnormal blood vessels may be small and cutaneous or mucosal (telangiectasia), with frequent complications of bleeding, or large and visceral (arteriovenous malformations [AVMs]), with additional risks that can lead to significant morbidity and even mortality. HHT can present in many different ways and can be difficult to recognize, particularly in younger patients in the absence of a known family history of disease or epistaxis, its most common manifestation. HHT is commonly diagnosed using the established Curaçao clinical criteria, which include (1) family history, (2) recurrent epistaxis, (3) telangiectasia, and (4) visceral AVMs. Fulfillment of 3 or more criteria provides a definite diagnosis of HHT, whereas 2 criteria constitute a possible diagnosis of HHT. However, these criteria are insufficient in children to rule out disease due to the age-dependent development of some of these criteria. Genetic testing, when positive, can provide definitive diagnosis of HHT in all age groups. Clinical course is often complicated by significant epistaxis and/or gastrointestinal bleeding, leading to anemia in half of adult patients with HHT. The management paradigm has recently shifted from surgical approaches to medical treatments aimed at control of chronic bleeding, such as antifibrinolytic and antiangiogenic agents, combined with aggressive iron replacement with intravenous iron. Guidelines for management of HHT, including screening and treatment, were determined by expert consensus and originally published in 2009 with updates and new guidelines in 2020.
Learning Objectives
Recognize signs and symptoms of HHT
Know when to test/screen patients for additional complications of the disease
Understand the approach to treatment of various manifestations of HHT
CLINICAL CASE
Presentation. A 17-year-old previously healthy girl presented to neurology with a complaint of intractable headaches and a history of possible prior concussions during sports. Magnetic resonance imaging (MRI) without contrast revealed some asymmetry of the cerebral vessels, and subsequent MRI with contrast and magnetic resonance arteriography revealed an arteriovenous malformation (AVM) involving the left perisylvian fissure and insular areas. This was not felt to be the cause of her headaches, and an electroencephalogram ruled out seizure activity. She was referred to neurosurgery for further management. The neurosurgeon noted telangiectasia on her fingertips and elicited a history of heavy recurrent nosebleeds since the age of 9 years. Further questioning revealed that her mother and brother also had frequent epistaxis and migraine headaches and that her mother had undergone a lung lobectomy for AVMs. At that time, the neurosurgeon informed the family that the patient likely had hereditary hemorrhagic telangiectasia (HHT).
Diagnosis. The patient was referred to our HHT center 20 months after seeing neurosurgery at the age of 18 years. She continued to have significant migraine headaches despite medication. She had nosebleeds approximately 1/wk, often gushing, lasting up to 15 minutes (Epistaxis Severity Score [ESS] 3.7). Physical examination confirmed telangiectasia on her fingers, lower lip, and tongue. Family history was confirmed, including distant cousins with brain and lung AVMs. The patient met Curaçao clinical criteria for definite HHT (+telangiectasia, +epistaxis, +AVM) at this visit even in the absence of a diagnosis in a first-degree relative. She underwent genetic testing to confirm the diagnosis, and a pathogenic variant in the endoglin gene ENG was identified.
Screening. Additional screening was performed to evaluate for lung AVMs. A delayed-bubble echo was positive, and subsequent computed tomography angiography of the chest revealed a left lower lobe AVM with a nidus measuring nearly 2×1×1 cm, with a feeding artery measuring 4 mm.
Treatment. She underwent embolization of her pulmonary AVM with dramatic improvement in her migraine headaches. At the age of 22 years, she underwent precision radiotherapy to multiple brain AVMs due to concern for progressive venous ectasia.
Follow-up. She continues to be followed in our HHT Center of Excellence at regular intervals to monitor for new or recanalized pulmonary AVMs (bubble echo every 3-5 years) and changes in epistaxis (ESS at every visit). She gets annual laboratory tests to screen for complications of liver AVMs (hepatic panel including γ-glutamyl transferase [GGT] to assess liver function and brain natriuretic peptide [BNP] for heart function) and anemia (complete blood count [CBC], ferritin, total iron binding capacity [TIBC], and reticulocyte count) related to nosebleeds or potential gastrointestinal (GI) bleeding. She takes antibiotics prior to dental procedures, including regular cleaning, for infection prophylaxis, due to her known lung AVMs as per HHT treatment guidelines. She is also followed in the cerebrovascular clinic for her cerebral AVMs.
Her family members also underwent screening for visceral vascular lesions given their clinical diagnoses of HHT once our patient was diagnosed. Her brother was found to have both multiple brain AVMs, which were treated with radiotherapy, and multiple lung AVMs, which were treated with coil embolization. Her mother was found to have additional lung AVMs that required embolization. Furthermore, her mother proceeded to develop severe iron deficiency anemia secondary to both severe epistaxis and GI bleeding and is taking maintenance therapy with systemic bevacizumab in addition to intermittent iron infusions to maintain ferritin more than 50 ng/mL.
Introduction
Hereditary hemorrhagic telangiectasia is the second most common inherited bleeding disorder, occurring in 1/5000 to 1/10,000 people.6 It is generally inherited in an autosomal dominant fashion but can occur de novo, including in mosaic form in some probands.7-11 Bleeding is the most common symptom, occurring from nasal, cutaneous, or GI telangiectases. Unlike bleeding disorders such as hemophilia or von Willebrand disease, bleeding in HHT is secondary to development of malformed blood vessels, which result in ectasia and increased fragility. This leads to localized bleeding from the abnormal vessels, rather than the general risk of bleeding and postsurgical bleeding associated with deficiencies of plasma coagulation factors or platelet dysfunction.
Diagnosis
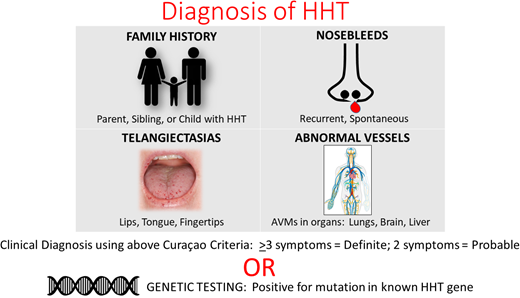
The Curaçao clinical diagnostic criteria, first formalized in 2000, are shown in Table 1.12 The first is epistaxis; to meet this criterion, nosebleeds should be recurrent and spontaneous. These may be gushing or dripping, as there is no requirement for severity.
While there is no widely accepted nosebleed frequency to meet this criterion, we generally use more than 4 per year with no other cause.5 A history of nighttime nosebleeds is particularly suggestive of HHT.
Telangiectases, or small bright red spots, typically occur at characteristic sites in HHT, including lips, oral cavity (particularly tongue), fingers, and nose (Figure 1). These telangiectasia can be tiny and appear like red freckles; they generally blanch with pressure, with rapid reperfusion upon release. Visceral lesions can also be present, including GI telangiectases and larger high-flow lesions, including (in order of frequency) liver, lung, brain, and spinal AVMs. These can be asymptomatic and may only be detected with screening. A family history of similar symptoms in a first-degree relative (parent, sibling, or child) is usually present even if they do not always carry a diagnosis of HHT. Underrecognition and delayed diagnosis is a considerable problem in HHT,13 and there is a need to increase awareness among health care providers.
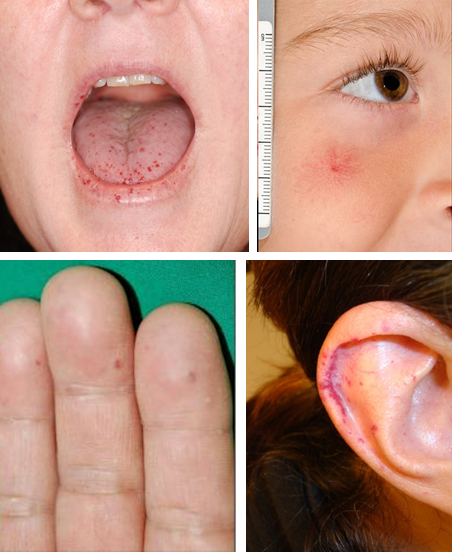
There is similarly no standard required telangiectasia count; in our centers, we use more than 4.5
Clinical criteria, validated in adults, may not be sufficient to rule out disease in children younger than 16 years.14 In 1 study by Pahl et al,14 the positive predictive value of the clinical criteria was extremely high in children (99% overall), but the negative predictive value was only 54% overall and even lower in children younger than 5 years. This is due to the age-dependent development of 2 of the criteria. Nosebleeds typically develop in the second decade of life and do not commonly require medical attention in childhood, although many patients do develop recurrent and/or severe epistaxis requiring medical and/or surgical interventions by middle age. Development of telangiectasia is also age dependent, typically noted in the second and third decades. Thus, even when a child has a parent with HHT (1 clinical criterion), in the absence of imaging performed to screen for visceral AVMs, affected children may not meet the diagnostic threshold for disease.
Genetic testing has become more available and affordable and can be used to make the HHT diagnosis.15 HHT-causing mutations have been identified in several genes in the transforming growth factor β pathway, with the majority of pathogenic variants found in ENG and ACVRL1. In 1 recent study, ENG and ACVRL1 mutations were found to comprise up to 96% of cases of “classic HHT” meeting strictly applied Curaçao criteria.16 SMAD4 gene mutations account for approximately 10% of ENG- and ACVRL1-negative cases of HHT, accounting for 1% to 2% of cases overall.16,17 SMAD4 also causes juvenile polyposis, resulting in GI polyps and a cancer predisposition syndrome,18 and has been implicated in aortopathy as well.19 Rarely, HHT cases meeting clinical criteria may be caused by GDF2 (BMP9) or RASA1.20 Overlap has been observed between HHT and capillary‑ malformation–AVM syndrome caused by EPHB4.21 In practice, most testing is now performed as part of multigene panels consisting of 5 to 6 genes (ENG, ACVRL1, SMAD4, RASA1, GDF2, and EPHB4).
Genetic testing is recommended to assist in establishing the diagnosis in individuals who do not meet Curaçao criteria or in those who are asymptomatic or minimally symptomatic, including young children. Genetic testing can also be used to identify the causative mutation in a family with clinically confirmed HHT or to establish a diagnosis in relatives of a person with a known causative mutation.15 However, current testing remains unable to identify a causative genetic change in up to 10% to 15% of patients with a clinical diagnosis.
Several laboratories now offer free family testing within a specified time period when a pathogenic variant is identified in 1 patient.
Guidelines have been established regarding the diagnosis, screening, and treatment of people living with HHT. The original guidelines were published in 2011 and covered diagnosis of HHT, epistaxis, cerebral vascular malformations, pulmonary AVMs, GI bleeding, and liver vascular malformations.15 The Second International guidelines were published in 2020, with updated guidelines for epistaxis, GI bleeding, and liver vascular malformations, as well as new guidelines on anemia and anticoagulation, pediatrics, and pregnancy and delivery.1
Clinical manifestations and management
Initial screening
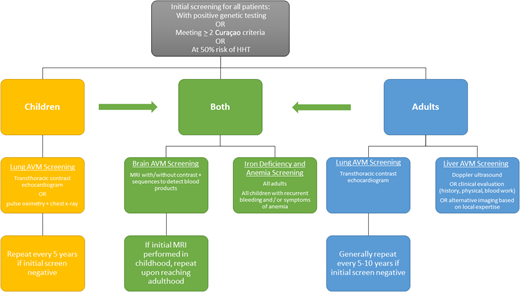
Once the diagnosis of HHT is made or suspected, all patients should undergo initial screening for potential complications. Although a long-time area of debate in the care of patients with HHT, recent pediatric care guidelines clarify that children require workup and screening as well as adults, because even though the outward clinical signs (telangiectasia) and symptoms (epistaxis) may so far be absent, pediatric patients can still have visceral AVMs that can cause morbidity and even mortality. Screening procedures are summarized in Figure 2 from the guidelines for pulmonary AVMs, liver vascular malformations, brain vascular malformations, and pediatric care.
Epistaxis
Epistaxis, the most prominent symptom in most people with HHT, can significantly interfere with daily activities, including work, school, and quality of life (QoL). The updated guidelines outline a number of recommendations for control of nosebleeds in a stepwise fashion.1 The ESS was previously established for use in HHT to facilitate measurement of nosebleeds based on a number of factors, including frequency, duration, amount of bleeding and presence of anemia, and interventions needed to control the bleeding.22 This score should be calculated at every visit and before and after any interventions to measure their impact (online calculator readily available at https://curehht.org/resource/epistaxis-severity-score/). ESS can be used to guide treatment of epistaxis, as shown in Figure 3. Although historically approaches were primarily surgical, medical therapies including antifibrinolytics (tranexamic acid, epsilon aminocaproic acid)23,24 and systemic antiangiogenics have become the mainstay of treatment strategies in recent years, as discussed in more detail below under “Anemia.”
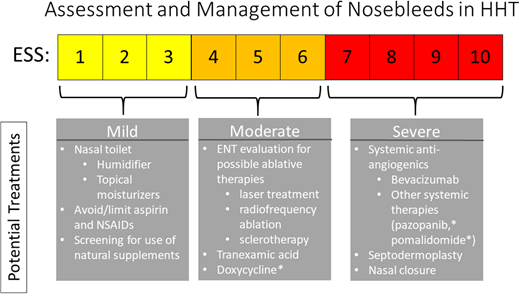
Epistaxis Severity Score (ESS)22 and management of epistaxis in HHT.1 *Therapy currently under investigation. ENT, ear, nose, and throat; NSAID, nonsteroidal anti-inflammatory drug.
GI bleeding
GI bleeding is also a common symptom in adults with HHT older than 50 years, reported in 13% to 30% of patients. Most GI telangiectasias occur in the stomach (46%-75%) and small bowel (56%-91%).25 GI bleeding can be highly morbid in adults, with chronic GI bleeding resulting in severe iron deficiency anemia requiring frequent intravenous iron replacement and/or red blood cell transfusions, with decreased QoL. GI bleeding is rarely significant in children except in those carrying the SMAD4 genotype and then is typically related to bleeding from polyps. GI bleeding should be suspected in the presence of iron deficiency with or without anemia, particularly when this is out of proportion to reported epistaxis. The first step in evaluation is generally referral to a gastroenterologist familiar with HHT for upper endoscopy, as stool occult blood tests are not reliable in the setting of ongoing epistaxis. Recommended treatment is determined based on the severity of GI bleeding and coexisting anemia; recommendations for the workup and treatment of GI bleeding can be found in Figure 4.
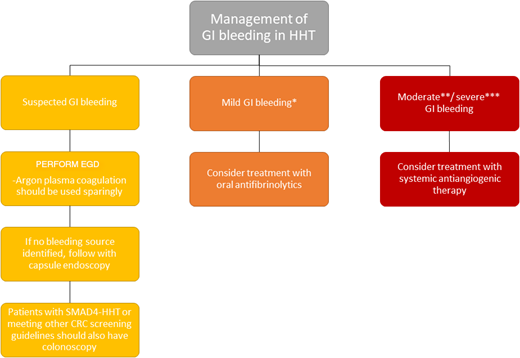
Guidelines for the management of GI bleeding in HHT.1 *Mild GI bleeding defined as meeting hemoglobin goals with oral iron. **Moderate GI bleeding defined as meeting hemoglobin goals with intravenous oral iron. ***Severe GI bleeding defined as not meeting hemoglobin goals despite adequate iron replacement or requiring blood transfusions. CRC, colorectal cancer; EGD, esophagogastroduodenoscopy.
Guidelines for the management of GI bleeding in HHT.1 *Mild GI bleeding defined as meeting hemoglobin goals with oral iron. **Moderate GI bleeding defined as meeting hemoglobin goals with intravenous oral iron. ***Severe GI bleeding defined as not meeting hemoglobin goals despite adequate iron replacement or requiring blood transfusions. CRC, colorectal cancer; EGD, esophagogastroduodenoscopy.
Anemia
Anemia can result from epistaxis, GI bleeding, or the combination of the two and is present in more than 50% of adults with HHT.26 Anemia, and even iron deficiency without anemia, should be managed aggressively to correct anemia and maintain adequate iron stores. In addition to well-known signs and symptoms of anemia, including fatigue, shortness of breath, lightheadedness, pallor, and cardiac stress, there have been additional associations reported in patients with HHT. In patients with liver AVMs, iron deficiency results in substantial increased risk of aberrant manganese deposition in the brain, leading to Parkinson-like neurologic symptoms.27 Paradoxically, an increased risk for thrombosis has been reported in HHT, which appears to correlate with iron deficiency.28 Such thrombotic events are generally treated with anticoagulation, with associated increased bleeding risk. While anticoagulation is not absolutely contraindicated in HHT, it can be quite difficult to balance bleeding risks and clotting risks in these patients. Complete recommendations for management of anemia and anticoagulation in HHT can be found in Figure 5.
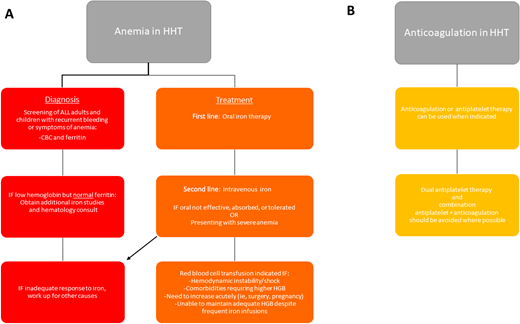
Guidelines for the management of anemia and anticoagulation in HHT.1 HGB, hemoglobin.
Our goal is to try to achieve and maintain ferritin above 50 ng/mL in the setting of ongoing recurrent bleeding.
For anemia resulting from epistaxis or GI bleeding refractory to local therapies and first-line medical treatments, systemic antiangiogenic therapies should be considered in combination with aggressive iron replacement. To date, the most widely used antiangiogenic agent in HHT is bevacizumab, a monoclonal anti–vascular endothelial growth factor antibody given as an intravenous infusion.29,30 Although systemic bevacizumab has not been studied in a prospective manner in HHT, it has become the de facto standard of care for severe/refractory anemia in HHT. The efficacy of the oral antiangiogenic agents pomalidomide (PATH-HHT trial; clinicaltrials.gov NCT03910244) and pazopanib (clinical trial opening soon) are being evaluated prospectively in clinical trials following promising data in pilot studies.31,32 Additional agents, including doxycycline33 (NCT03397004, NCT04167085) and tacrolimus (NCT04646356), are also being actively evaluated.
Visceral AVMs
Visceral AVMs, although less common than bleeding symptoms, can also cause significant morbidity and mortality if undetected and/or untreated. Therefore, it is important to be aware of potential signs and symptoms of AVMs and follow current screening guidelines. In doing so, individuals with HHT who undergo recommended screening and treatment at an HHT Center of Excellence can expect a normal life span.34,35
Brain vascular malformations seen in HHT include AVM, arteriovenous fistula (AVF), vein of Galen malformations, cerebral cavernous malformations, developmental venous anomalies, and capillary telangiectasia. Of these, AVF and AVM are considered highest risk for complications. Symptoms of AVM vary by location and can include seizure, headache, or, in the case of rupture, hemorrhagic stroke; however, most brain AVMs are asymptomatic and found on routine screening. Management of brain vascular malformations, once identified, is pursued collaboratively with neuroradiology, neurology, neurosurgery, and/or neurointerventional radiology. Treatment options for high-risk lesions include embolization by a skilled neurointerventional radiologist, resection by a neurosurgeon, or focal radiation (gamma knife or proton) administered by a radiation oncologist; choice of therapy will depend on risk of the lesion, size, and location, including considerations of accessibility and eloquence.
Lung AVMs can be asymptomatic or may cause a number of signs and symptoms. These include chronic manifestations such as fatigue, shortness of breath, hypoxia, asthma-like symptoms poorly responsive to β-agonists, migraine headaches, or dramatic and life-threatening presentations such as paradoxical embolization leading to stroke, transient ischemic attack, or cerebral abscess. AVM rupture causing massive hemoptysis and/or spontaneous hemothorax is a rare presentation. Cerebral abscess risk has been strongly associated with dental work, including routine cleaning, and antibiotic prophylaxis should be administered prior to dental work to all patients with known lung AVMs and those who have not yet undergone lung AVM screening to prevent this complication. Many HHT centers also recommend antibiotic prophylaxis for other “dirty” procedures that could potentially introduce bacteria into the bloodstream, including colonoscopy and other GI/GU (genitourinary) procedures, but this is not yet standardized. Treatment of lung AVMs is generally carried out by interventional radiologists using coil embolization. The decision to treat lung AVMs is based on size of the AVM nidus and diameter of the feeding vessels (typically >2-2.5 mm diameter depending on the center) but may also be considered for slightly smaller AVMs based on symptoms. After treatment, continued monitoring is necessary to rule out recanalization or formation of new AVMs; intervals vary on a case-by-case basis. An update to the pulmonary AVM guidelines is planned in the near future.
Liver AVMs are the most common visceral AVM found in HHT, but they are often asymptomatic and have been historically hard to treat. The updated guidelines recommend screening, as it may be possible to pick up sequelae of the hepatic AVMs earlier this way, including high-output heart failure, pulmonary hypertension, portal hypertension, and encephalopathy. When present, liver AVMs should be followed by a hepatologist but, depending on the manifestations, may require cardiology and/or pulmonology or pulmonary hypertension expertise. Treatment options for hepatic AVMs have been limited, as embolization often leads to further liver complications. Current approaches include orthotopic liver transplant or systemic medical therapies such as bevacizumab, reviewed in more detail above in the “Anemia” section.
Pregnancy and childbirth
Genetic counseling is recommended preconception to discuss risks of transmission and/or prenatally to explore multiple options for genetic testing of the child, from preimplantation, to in utero, to postnatal options such as cord blood. Guidelines for pregnancy and delivery were also developed, including screening recommendations for AVMs, outlined in Figure 6. Women who have not been screened recently or who have known lung AVMs and/or those with high-risk brain vascular malformations should be followed by a multidisciplinary team, including a maternal-fetal medicine specialist, at a tertiary care center. Lung AVMs in need of treatment should be embolized in the second trimester if possible and patients counseled that hemoptysis is an emergency, based on increasing evidence of maternal morbidity and even mortality during pregnancy and childbirth,36 due at least in part to increased risk of lung AVM rupture.37,38
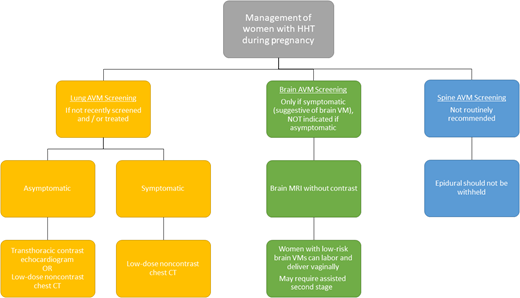
Guidelines for pregnancy and delivery in HHT.1 CT, computed tomography; VM, vascular malformation.
Guidelines for pregnancy and delivery in HHT.1 CT, computed tomography; VM, vascular malformation.
Conclusion
Patients with HHT can initially present to any of several different medical specialists, depending on their presenting symptoms, and underrecognition of the disease risks diagnostic delays and increased morbidity. It is important that potential providers have a high index of suspicion in the presence of the Curaçao clinical criteria for HHT. These patients can present to hematologists in many ways: with anemia and iron deficiency (due to bleeding from nasal or GI telangiectases), with polycythemia (as a normal physiologic response in the setting of pulmonary AVMs), or for systemic therapies for other complications of HHT (severe anemia requiring significant iron/blood replacement or pulmonary hypertension or high-output cardiac failure related to liver AVMs).39 Where HHT Centers of Excellence exist, currently at 30 sites in North America, the center's director or codirector will likely provide a medical home for these often complicated patients; however, no such center exists in most of the medical centers across the country. Elsewhere, hematologist-oncologists may find themselves in the role of primary provider, particularly as more targeted medical therapies become available as, to date, most of these drugs have been used previously in the treatment of cancers. Given this and the relatively high frequency of HHT as a hereditary bleeding disorder, an understanding of modern HHT management is crucial for the practicing hematologist.
Conflict-of-interest disclosure
Adrienne M. Hammill: none related to this paper. But AH has clinical trials (for other diseases) with novartis, cerecor, Venthera, Merck; had study drug provided by Pfizer; serves as a consultant for Novartis.
Katie Wusik: none reported.
Raj S. Kasthuri: none reported.
Off-label drug
Adrienne M. Hammill: All drugs listed here are off label. None has FDA approval. They are not attributable to a single author.
Katie Wusik: All drugs listed here are off label. None has FDA approval. They are not attributable to a single author.
Raj S. Kasthuri: All drugs listed here are off label. None has FDA approval. They are not attributable to a single author.