

Abstract
The development of novel cellular therapies and bispecific T-cell-engaging antibodies is occurring at breakneck speed in multiple myeloma (MM). While groundbreaking, these agents have their unique logistical and toxicity issues and currently do not represent a curative approach. In this context, there continues to be an urgent need to develop novel, off-the-shelf immunotherapy approaches to add to the armamentarium. This article explores novel agents being investigated in combination with standard immunomodulatory drugs as well as next-generation cereblon E3 ligase modulators. These novel agents include drugs being repurposed from their use in other diseases as well as novel monoclonal antibodies. In addition, agents under development such as immunocytokines, immunotoxins, and natural killer-cell activators/engagers are reviewed. These novel therapeutic strategies hold the promise of countermanding the immunosuppressive tumor microenvironment, leading to enhanced anti-MM activity.
Learning Objectives
Learn about novel IMiD-based combinations and CELMoDs being tested in R/R MM
Recognize the developing therapeutic landscape of immunocytokines, immunotoxins, and NK-cell activators/engagers in MM
CLINICAL CASE
A 64-year-old woman with immunoglobulin G (IgG) lambda relapsed/refractory multiple myeloma (R/R MM) received commercial idecabtagene vicleucel 6 months ago and is now experiencing biochemical relapse. She had previously received 5 prior lines of therapy, including prior autologous stem cell transplant (ASCT), and her disease is refractory to lenalidomide, pomalidomide, bortezomib, carfilzomib, daratumumab, and belantamab mafodotin. Her chimeric antigen receptor (CAR) T-cell course was complicated by cytokine release syndrome and neurotoxicity, and she wishes to avoid any subsequent therapies that could be associated with those adverse events (AEs). What novel non–CAR T-cell/non–bispecific antibody immunotherapy approaches are currently under investigation?
Introduction
Novel immunotherapies such as CAR T cells and bispecific T-cell–engaging antibodies show remarkable promise for MM. However, given the current logistical constraints of commercial CAR T-cell products, as well as the potential logistical issues with bispecific antibodies that may limit access to these therapies to certain populations, there continues to be a need for alternative approaches. With the remarkable success of small-molecule agents such as immunomodulatory drugs (IMiDs) and proteasome inhibitors, the ongoing substantial focus on developing novel small-molecule agents for MM is not surprising. In addition, the field is exploring the therapeutic potential of novel antibody-based approaches and natural killer (NK)-cell activators/engagers. In this educational session, we discuss the most recent research investigating these novel non–CAR T-cell/bispecific-based approaches for the treatment of R/R MM.
IMiDs plus novel agent combinations
The IMiDs lenalidomide and pomalidomide have helped transform the management of MM. Lenalidomide is a current standard of care in the newly diagnosed (ND) and maintenance settings, while pomalidomide is exclusively used in the R/R setting. The pleiotropic effects of these agents, which include both direct cytotoxic effects as well as a myriad of effects on the immune microenvironment, coupled with generally tolerable side-effect profiles, have led to numerous combination-based studies. Substantial research is focused on determining the mechanisms of resistance to these agents and whether these resistance mechanisms can be overcome via novel combinations. In this section an overview of some of the most recent IMiD plus novel agent combination strategies are provided, including those involving small molecules (Table 1) and antibody-based therapies (Table 2). In several cases, agents being used to treat other malignancies or diseases are being repurposed for the treatment of MM. Other ongoing studies are investigating IMiD-based combinations with agents such as lisaftoclax (BCL2 inhibitor), KRT-232 (MDM2 inhibitor), or novel histone deacetylase inhibitors (eg, ricolinostat, ACY-241, chidamide) (Table 1). Not discussed in this chapter but included in Table 2 are a number of ongoing studies evaluating IMiDs with bispecific antibodies.
The Bruton tyrosine kinase (BTK) inhibitor ibrutinib is now widely used in the treatment of several B-cell malignancies. BTK is expressed in MM cells, and it may modulate the bone marrow (BM) microenvironment by decreasing cytokine/chemokine secretion and suppressing myeloid-derived suppressor cell activity.1 While single-agent ibrutinib led to an overall response rate (ORR) of 0% in patients with R/R MM (64% lenalidomide refractory),2 subsequent efforts focused on combining the agent with IMiDs. A phase 1 study of ibrutinib-lenalidomide- dexamethasone reported an ORR of 7% with a clinical benefit rate (≥ minimal response) of 80%. Notably, 54% of the patients were refractory to lenalidomide.3 An ongoing phase 1 study is evaluating the combination in patients with at least 1 prior line of therapy (eligible patients must not have progressed on full-dose lenalidomide but could have experienced disease progression on lenalidomide maintenance) (Table 1). A phase 1 study of ibrutinib in combination with pomalidomide-dexamethasone was completed, and the sponsor chose not to pursue the phase 2 component (NCT02548962).
The Janus kinase (Jak) inhibitor ruxolitinib has a number of immunomodulatory properties that appear relevant for MM. Recent work has shown that ruxolitinib can decrease PD-L1 expression and decrease expression of a variety of genes implicated in resistance to lenalidomide.4,5 A phase 1 study of ruxolitinib in combination with lenalidomide and methylprednisolone reported an ORR of 38% in a patient population in which 93% were lenalidomide refractory.6
Leflunomide was approved for the treatment of rheumatoid arthritis by the US Food and Drug Administration in 1998. Leflunomide is metabolized to teriflunomide, which inhibits dihydroorate dehydrogenase, an enzyme involved in pyrimidine biosynthesis. While earlier studies suggested that leflunomide induced cytotoxicity in MM cells in part related to the disruption of pyrimidine biosynthesis, more recent work has indicated that this agent downregulates c-Myc expression by directly inhibiting the PIM family of serine/threonine kinases.7 Preclinical studies showed the anti-MM activity of leflunomide but only in immune-competent mice.7 Increases in T-cell activation markers (LAMP-1, CD69) with a decrease in the T-cell exhaustion marker CTLA4 were observed in treated animals.7 In addition, the combination of lenalidomide with leflunomide prolonged survival in a mouse model of MM.7 Subsequently a phase 1 study of single-agent leflunomide was conducted in patients with R/R MM. No objective responses were reported; however, 9 in 11 patients achieved stable disease.8 Currently, this agent is being investigated in patients with smoldering MM as well as in combination with pomalidomide-dexamethasone in patients with R/R MM.
Vactosertib is a transforming growth factor beta 1 (TGFB1) receptor antagonist, and as there are data connecting increased TGFB1 secretion by MM cells with impaired immune surveillance, studies have been conducted evaluating the potential therapeutic use of vactosertib in MM. Preclinical studies with vactosertib demonstrated single-agent activity in a mouse MM model as well as in primary patient samples.9 A phase 1 study was conducted evaluating the combination of vactosertib with pomalidomide (no steroids) and demonstrated the safety of this combination along with a 6-month progression-free survival (PFS) rate of 80% (n = 15).10
Tasquinimod is an agent that targets myeloid-derived suppressor cells via the inhibition of S100A9. While thus far it has primarily been investigated in the setting of prostate cancer, preclinical studies in MM revealed single-agent activity as well as in combination with lenalidomide.11 No direct cytotoxic effect of this agent was observed against MM cells in vitro, suggesting that the mechanism of action is via modulation of the tumor microenvironment.11 A phase 1 study of single-agent tasquinimod in R/R MM was completed and this agent is currently being evaluated in combination with ixazomib-lenalidomide- dexamethasone (NCT04405167).
While several phase 3 trials studying IMiDs in combination with anti-programmed cell death 1 protein inhibitors such as pembrolizumab and nivolumab failed,12–14 there continues to be interest in investigating the therapeutic potential of alternative checkpoint inhibitors. Several ongoing clinical trials are evaluating combinations of IMiDs with novel checkpoint inhibitors such as anti-LAG3 and anti-TIGIT monoclonal antibodies (mAbs) (Table 2). There is also interest in targeting CD47, often referred to as the macrophage “don't eat me” signal, and recent/ongoing studies are exploring magrolimab- or lemzoparlimab-based combinations in R/R MM. Whether the toxicity issues encountered with IMiDs and anti-programmed cell death 1 inhibitors will also be observed with any of these novel immune-targeting mAbs remains to be determined,15 but it is notable that the lemzoparlimab-based study was recently terminated by the sponsor for as yet undisclosed reasons while unexpected serious AEs were reported with magrolimab in other malignancies.
CELMoDs
Iberdomide (CC-220) is an orally available agent that, similar to lenalidomide and pomalidomide, binds to the cereblon E3 ubiquitin ligase complex and thus is considered to be a CELMoD (cereblon E3 ligase modulator). Iberdomide more potently binds cereblon, thus leading to greater degradation of Ikaros and Ailos in cell culture studies compared to lenalidomide or pomalidomide.16 Preclinical studies demonstrated the activity of iberdomide in lenalidomide- or pomalidomide-refractory MM cells.17 A phase 1b/2a dose-escalation study of iberdomide in R/R MM was conducted.18 Iberdomide (daily on days 1-21 of a 28-day cycle) was administered in combination with weekly dexamethasone. The median number of prior regimens was 5 (2-12), with 100% receiving lenalidomide and 69% receiving prior pomalidomide. Dosing started at 0.3 mg, and the recommended phase 2 dose (RP2D) was determined to be 1.6 mg. The most common grade 3 to 4 AEs were neutropenia (26%), thrombocytopenia (11%), and neuropathy (2%). The ORR was 31% while 88% achieved stable disease or better (disease control rate).18 The results from the dose-expansion phase of this study showed an ORR of 28% with a clinical benefit rate of 36% and a disease control rate of 79% (n = 107). In patients who had previously received anti–B-cell maturation antigen (BCMA) therapy, the ORR was 25% (n = 24).19 Mass cytometry analysis of BM aspirate samples from patients treated in the phase 1b/2a study revealed marked changes in the tumor microenvironment, including a reduction in naive and regulatory B cells, a reduction in CD4+ T cells with an increase in CD8+ T-cells, a shift toward the cytotoxic effector-memory phenotype of T cells, a decrease in CD8+ T cells expressing inhibitory checkpoints (TIGIT, KLRG1), and an increase in NK cells (including activated NK cells) with a decrease in TIGIT+ NK cells.20
Clinical trials investigating iberdomide in combination with other standard agents are ongoing (Table 3). Preliminary results from a phase 1/2 study evaluating iberdomide in combination with either daratumumab-dexamethasone or bortezomib- dexamethasone have been reported.21 The RP2D of iberdomide in both combinations was also determined to be 1.6 mg daily. The rate of grade 3 to 4 neutropenia in the iberdomide-daratumumab-dexamethasone cohort was 67% and 26% in the iberdomide-bortezomib-dexamethasone cohort. The rates of diarrhea (all grades) were 26% (iberdomide-daratumumab-dexamethasone) and 35% (iberdomide-bortezomib-dexamethasone). No patients developed thrombotic events. Antithrombotic prophylaxis is mandatory in all ongoing studies with iberdomide. Other iberdomide-based combination trials include ixazomib-iberdomide-dexamethasone, iberdomide-cyclophosphamide- dexamethasone, and iberdomide-carfilzomib-dexamethasone, as well as iberdomide in combination with EOS884448-GSK4428859A, an anti-TIGIT monoclonal antibody. In addition, several studies are evaluating iberdomide as maintenance therapy post ASCT (Table 3).
Mezigdomide (CC-92480) is another potent CELMoD. This agent has a significantly higher degradation efficiency compared to either lenalidomide or pomalidomide.22 The phase 1 study of CC-92480 in heavily treated R/R MM patients (median prior lines = 6; half had triple-class refractory disease) in combination with dexamethasone showed an ORR of 55% at the RP2D (1.0 mg/d for 21 to 28 days).23 Grade 3/4 treatment-emergent AEs (TEAEs) were reported in 88% of patients, with the most frequent grade 3 and 4 AEs including neutropenia (53%), infections (30%), anemia (29%), and thrombocytopenia (17%). Correlative studies have shown Ikaros/Aiolos degradation in peripheral T cells in a dose-dependent manner at doses greater than or equal to 0.6 mg and showed substrate recovery during drug holidays (full recovery with ≥7-day breaks).24 Preliminary results have demonstrated the safety and feasibility of combining mezigdomide with bortezomib-dexamethasone,25 and ongoing studies include other mezigdomide-based combinations, including carfilzomib-dexamethasone, elotuzumab-dexamethasone, and isatuximab (or daratumumab)-dexamethasone (Table 3). In addition, more novel combinations such as mezigdomide-dexamethasone with tazemetostat (an EZH2 inhibitor), BMS-986158 (a BET inhibitor), or trametinib (a MEK inhibitor) are also planned (NCT05372354).
Immunocytokines and immunotoxins
The US Food and Drug Administration–approved anti-CD38 mAbs daratumumab and isatuximab have found widespread use in the R/R setting as well as increasing use in the ND setting. The predominant mechanisms of action for these agents include direct induction of apoptosis, antibody-dependent cell-mediated cytotoxicity (ADCC), complement-dependent cytotoxicity, antibody-dependent cellular phagocytosis, and inhibition of CD38 ectoenzyme activity, although a variety of immunomodulatory effects have been described as well.26,27 As alternatives to “naked” mAbs, newer drug development efforts have focused on the modification of anti-CD38 mAbs, thus leading to the generation of immunocytokines and immunotoxins (Figure 1).
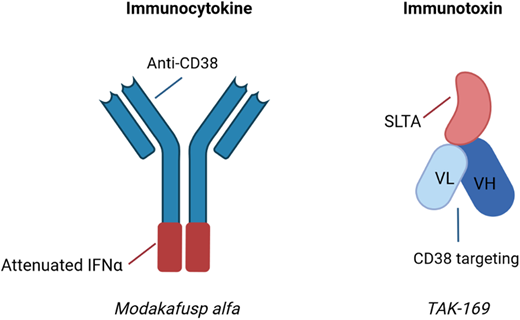
Examples of immunocytokines and immunotoxins undergoing clinical trial evaluation in MM. Immunocytokines are antibody-cytokine fusion proteins, while immunotoxins are fusion proteins containing antibody fragments and a biologic toxin. SLTA, Shiga-like toxin A subunit; VH, heavy chain variable region; VL, light chain variable region. Created with BioRender.com.
Examples of immunocytokines and immunotoxins undergoing clinical trial evaluation in MM. Immunocytokines are antibody-cytokine fusion proteins, while immunotoxins are fusion proteins containing antibody fragments and a biologic toxin. SLTA, Shiga-like toxin A subunit; VH, heavy chain variable region; VL, light chain variable region. Created with BioRender.com.
Modakafusp alfa (formerly known as TAK-573) is an example of an immunocytokine. This agent was designed as a delivery system of interferon alpha (IFNA) 2b to CD38+ cells. Modakafusp is a recombinant humanized IgG4 anti-CD38 mAb that is fused to an attenuated IFNA protein. While IFNA was investigated as an anti-MM agent decades ago due to its cytotoxic and immunomodulatory properties, the systemic toxicity of the agent limited its use.28 The hypothesis underlying the novel fusion protein is that the systemic toxicity is limited as a consequence of both reduced binding affinity to the IFNA receptor as well as an increased localized concentration at CD38+ target cells. In vitro studies demonstrated that modakafusp displayed 10 000-fold increased specificity for CD38+ cells while being approximately 6000-fold less toxic to normal BM cells compared to native IFNA.29 Preclinical studies showed synergy with standard agents such as lenalidomide and bortezomib.29 Due to the limited Fc functionality of this fused protein, it is thought to be unlikely to induce ADCC. This agent activates innate and adaptive immune cells, as well as induces apoptotic signals to the MM cells via signaling through the IFNA receptor.30 Interestingly, modakafusp binds to an epitope of CD38 that is unique relative to the commercially available anti-CD38 mAbs.31 A first-in-human phase 1 study in patients with R/R MM has shown that a dose of 1.5 mg/kg (intravenously) every 4 weeks resulted in an ORR of 38% in patients with a median of 7 prior lines of therapy, as well as an ORR of 38% in patients refractory to anti-CD38 mAb therapy.30 The most notable AEs included cytopenias (76% thrombocytopenia, 69% neutropenia, 66% anemia) and infusion-related reactions (31%).30 Currently, a randomized phase 2 study is evaluating 2 different dose levels (flat dosing of 120 or 240 mg every 4 weeks) in R/R MM.
TAK-169 is considered an immunotoxin because it is an engineered protein containing a deimmunized form of the ribosome-inactivating Shiga-like toxin A subunit (SLTA) fused to an antibody fragment that specifically targets the CD38 cell surface receptor.32 This agent is internalized by CD38-expressing cells, leading to cell death via the irreversible inhibition of protein synthesis. Ex vivo studies using primary BM samples showed lysis of the MM cells, with the highest amount of lysis in samples from ND or daratumumab-naive R/R patients.33 A phase 1 study in patients with R/R MM or non-Hodgkin lymphoma is ongoing (NCT04017130). An initial report after 4 patients were enrolled at the starting dose noted 1 patient experiencing a TEAE of asymptomatic grade 2 myocarditis that was reversible.34 A number of other immunotoxins have undergone preclinical evaluation thus far, targeting a variety of tumor antigens (eg, CD38, CD138, BCMA) and involving a number of toxins (eg, Shiga-like toxin A subunit SLTA, pseudomonas exotoxin, saporin, ricin).35
NK-cell activators and engagers
Determining how to effectively harness the anti-MM activity of NK cells has been an active area of research for decades. While many ongoing studies involve genetic engineering of NK cells (eg, CAR NK cells), interest in alternative strategies by which to enhance the activity of endogenous NK cells continues. Earlier efforts included mAbs targeting killer immunoglobulin-like receptors (KIRs; eg, IPH2101, 1-7F9, lirilumab) and NK group 2 member A (monalizumab). Currently, no ongoing studies are investigating these agents in MM.
An alternative strategy involves agents that enhance IL-15 activity, as IL-15 plays a key role in NK development and survival. ALT-803 is an IL-15 superagonist fusion protein that was evaluated in a phase 1 study in R/R MM; however, the results have not been disclosed (NCT02099539). A study using ALT-803 in patients relapsing after allogeneic stem cell transplant did demonstrate that this agent increased NK and CD8+ T-cell numbers and function.36 NKTR-255 is a polymer-conjugated IL-15 receptor agonist reported to have synergistic activity with daratumumab in preclinical models.37 Currently, this agent is being examined in a phase 1 study either as a single agent or in combination with daratumumab in R/R MM (NCT04136756).
Finally, an exciting emerging group of therapeutic agents are the bispecific and trispecific killer engagers (BiKEs and TriKEs), as well as the antibody-recruiting molecules (ARMs) (Figure 2). BiKEs include 2 linked single-chain variable fragments that recognize the tumor antigen (eg, BCMA) and an NK cell activation receptor (eg, NKp30, NK group 2 member A, CD16). TriKEs also incorporate another component, such as IL-15, to further enhance NK cell activation. Half-life extended nanobody-based BiKEs are under development.38 ARMs contain 2 linked termini; 1 terminus binds to a tumor antigen, while the other binds endogenous IgG antibodies regardless of the antigen-binding specificity.39 This leads to the opsonization of tumor cells with endogenous IgG, resulting in tumor lysis via NK cell–mediated ADCC.39 Preclinical studies have demonstrated the potential of BiKEs, TriKEs, and ARMs,39–41 and a clinical trial evaluation is the next step.
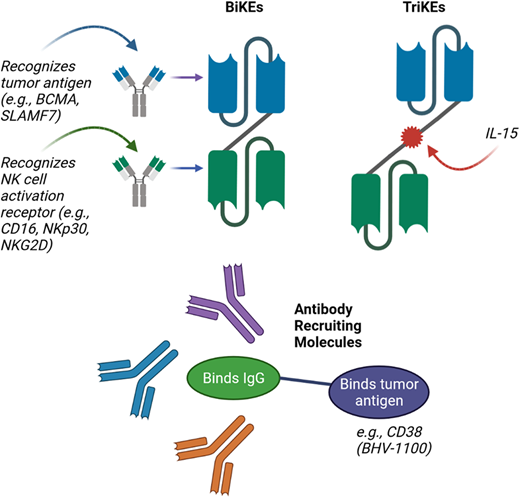
Novel approaches for improving NK-cell–mediated targeting of myeloma cells. Bispecific killer cell engagers (BiKEs) are composed of 2 antibody fragments that recognize tumor antigens and NK-cell activation receptors, while trispecific killer cell engagers (TriKEs) also include a component such as IL-15 to further enhance NK-cell activation. Antibody-recruiting molecules (ARMs) contain a terminus that binds endogenous IgG antibodies and is linked to a second terminus that binds tumor antigen, thus leading to opsonization of tumor cells with endogenous IgG and resulting in NK-cell–mediated ADCC.
Created with BioRender.com
Novel approaches for improving NK-cell–mediated targeting of myeloma cells. Bispecific killer cell engagers (BiKEs) are composed of 2 antibody fragments that recognize tumor antigens and NK-cell activation receptors, while trispecific killer cell engagers (TriKEs) also include a component such as IL-15 to further enhance NK-cell activation. Antibody-recruiting molecules (ARMs) contain a terminus that binds endogenous IgG antibodies and is linked to a second terminus that binds tumor antigen, thus leading to opsonization of tumor cells with endogenous IgG and resulting in NK-cell–mediated ADCC.
Created with BioRender.com
CLINICAL CASE (Continued)
A discussion is held with the patient about the many promising approaches currently under investigation, including the use of IMiDs with novel agents, CELMoDs, immunocytokines, and NK-cell activators/engagers. As all of these approaches involve modulation of the immune system in some manner, off-target effects are possible. She expresses interest in learning more about the clinical trial options available at your institution.
Conflict-of-interest disclosure
Sarah A. Holstein: consultancy: Bristol Myers Squibb, Celgene, Janssen, Genentech, Oncopeptides, Sanofi, Secura Bio, Takeda; research funding: Oncopeptides.
Off-label drug use
Sarah A. Holstein: by definition, everything that is discussed in this chapter is currently experimental and undergoing clinical trial and is therefore off-label.

