Abstract
Acute myeloid leukemia (AML) secondary to antecedent hematologic disorder or prior therapeutics for cancer represent a diverse group of leukemias often associated with inferior outcomes. Conventional therapy with cytarabine-based chemotherapy has been the mainstay of care for the past 30 years with disappointing overall outcomes. Novel therapies, including liposomal cytarabine/daunorubicin, and venetoclax-based therapies have emerged as options in recent years based on studies showing improvement in outcomes over standard-of-care therapies. Despite these advances, mutations in TP53 are associated with inferior response to both therapies and represent an area of unmet clinical need. Novel strategies with immune-targeted therapies such as CD47 monoclonal antibodies appear active in early-phase studies, but randomized studies have yet to report outcomes leading to approval. Allogeneic transplant remains the only known curative therapy for many of these cases. Nonetheless, pretransplant high-risk molecular features of secondary AML are associated with inferior outcome despite transplantation. An optimal approach to secondary AML is yet to be determined.
Learning Objectives
Secondary acute myeloid leukemia (AML) encompasses both therapy-related myeloid neoplasms and antecedent myelodysplastic syndrome and/or myeloproliferative neoplasm
Favorable risk secondary AML is rare but can be managed like de novo favorable risk disease
Allogeneic stem cell transplant remains the preferred option in first complete remission for intermediate- and unfavorable-risk disease
Venetoclax and hypomethylating agent combination may be used as a bridge to transplant or destination therapy
Emerging therapies may further expand treatment options for this historically older, frailer population with complex genetic changes and TP53 mutations
Introduction
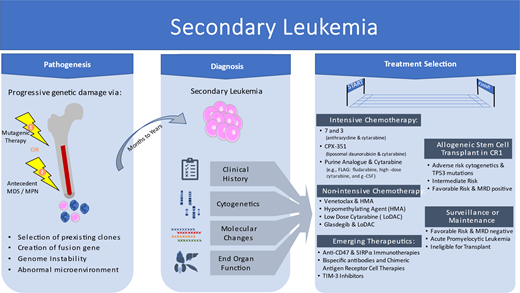
Secondary acute myeloid leukemia (AML) has been historically divided into 2 categories based on known risks for development of disease from either (1) antecedent myeloid neoplasm such as myelodysplastic syndrome (MDS) or myeloproliferative neoplasm (MPN) or (2) exposure to ionizing radiation and/or cytotoxic chemotherapy (Table 1). In population-based cohort series, secondary AML accounts for 20% to 30% of all AML cases.1,2 The previous World Health Organization (WHO) 2016 classification included the separate categories of AML with myelodysplastic changes (AML-MRC) and therapy-related myeloid neoplasms (tMNs), which were the formal categories of secondary AML.3 The WHO classification of tMN was subdivided into therapy-related acute myeloid leukemia and therapy-related myelodysplastic syndrome. The fact that both MDS and AML are included in the single category of “therapy-related myeloid neoplasm” emphasizes that regardless of precise blast count above or below 20%, similar biology, pathogenesis, and risk of rapid progression with poor prognosis guide the need for treatment and transplant evaluation for most patients.
The latest editions of the WHO classification4 and the International Consensus Classification5 further emphasize disease biology and genetic features while softening the boundary between MDS and AML. The WHO classification now specifically eliminates blast cutoffs for most AML types with defining genetic alterations but retains a 20% blast cutoff to delineate MDS from AML in cases lacking such genetic alterations. AML-MRC has been updated to AML, myelodysplasia related (AML-MR). AML-MR harbors specific mutational or cytogenetic abnormalities and can no longer be defined by morphology alone. Either the presence of 1 or more molecular and cytogenetic abnormalities or a history of MDS/MPN is required for a diagnosis of AML-MR. Further complicating these distinctions, patients with aplastic anemia or inherited bone marrow failure syndromes may also develop AML, particularly if dominant clones emerge. Far more commonly, patients have had prior cytotoxic therapy or known MDS/MPN, or they can be classified into AML-MR based solely on the presence of myelodysplasia-related chromosomal or mutational findings. Meanwhile, tMN has been replaced by myeloid neoplasm post–cytotoxic therapy, still requiring a documented history of chemotherapy treatment or large-field radiation therapy for an unrelated neoplasm.
Within both categories of AML-MR and AML, post–cytotoxic therapy (AML-pCT), similar chromosomal and molecular abnormalities are found, suggesting a common final pathway. Secondary leukemia from antecedent MDS/MPN appears to share a typical evolutionary pathway, with several genes distinguishing secondary AML from de novo AML with high specificity.6 Secondary leukemias after cytotoxic therapy may evolve through a similar MDS evolutionary pathway or acquire TP53 or other driver mutations directly from chemotherapy, radiation, or immunosuppression. Common cytogenetic and molecular features reflect this variability, with 50% of patients with AML-pCT having poor-risk cytogenetics. The most frequent molecular aberration is TP53 (33%), and only 15% of patients with AML-pCT present with favorable-risk fusion genes (RUNX1::RUNX1T1, CBFB::MYH11, PML::RARA). Age-related increase in clonal hematopoiesis7 may predispose patients to develop secondary leukemia as an accretion of mutations leads to myeloid neoplasms, especially under the selective pressure of chemotherapy.8
Therapy-related myeloid neoplasms may arise after a variety of treatments, with classic examples being alkylating agents leading to AML-pCT with antecedent MDS, with abnormalities of chromosome 5 and/or 7 or topoisomerase II inhibitors leading to KMT2A gene rearrangements at 11q23. Beyond recurring gene or chromosomal rearrangements, other therapies can lead to leukemia by selecting preexisting clones resistant to chemotherapy, with recent novel therapy examples including poly (ADP-ribose) polymerase inhibitors and lenalidomide (see Figure 1).1,9
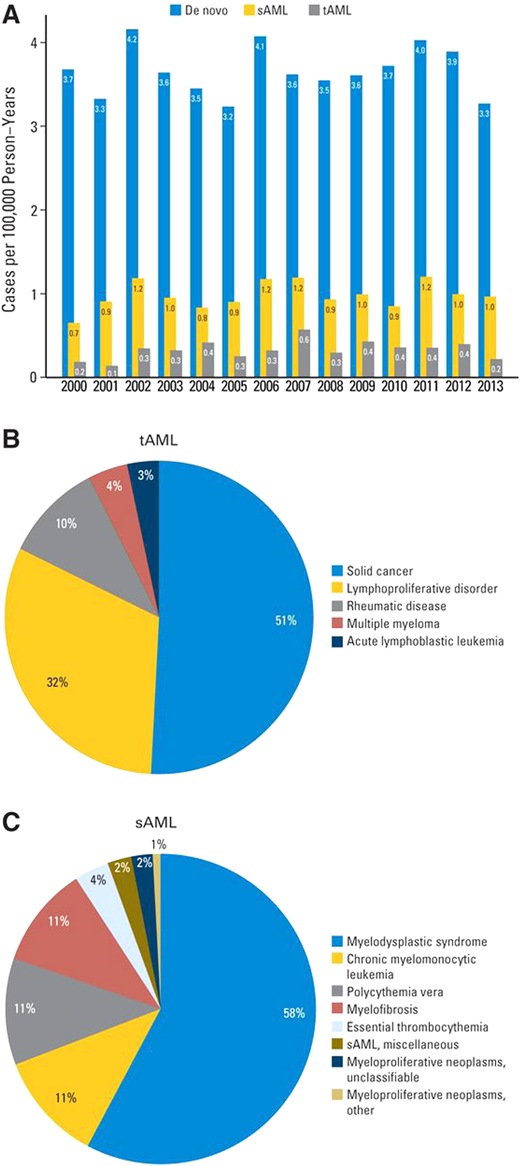
Descriptive data by AML type. (A) Incidence of de novo AML, secondary AML (sAML), and therapy-related AML (tAML) in Denmark by year of diagnosis. Distribution of previous disease in (B) 203 patients with tAML and (C) 603 patients with sAML. Originally published in Østgård LSG, Medeiros BC, Sengeløv H, et al. Epidemiology and clinical significance of secondary and therapy-related acute myeloid leukemia: a national population-based cohort study. J Clin Oncol. 2015;33(31):3641-3649.1
Descriptive data by AML type. (A) Incidence of de novo AML, secondary AML (sAML), and therapy-related AML (tAML) in Denmark by year of diagnosis. Distribution of previous disease in (B) 203 patients with tAML and (C) 603 patients with sAML. Originally published in Østgård LSG, Medeiros BC, Sengeløv H, et al. Epidemiology and clinical significance of secondary and therapy-related acute myeloid leukemia: a national population-based cohort study. J Clin Oncol. 2015;33(31):3641-3649.1
Outcomes in both AML-MR and AML-pCT appear similar across genetic subsets. Rare subsets of favorable-risk treatment- related disease such as AML with inv16 or t(8;21) and acute promyelocytic leukemia t(15;17) appear to have diminished overall survival compared with de novo AML. However, the impact of confounding from the antecedent malignancy or advanced age is often not examined in retrospective studies comparing outcomes in secondary AML to de novo AML. Clinically, diagnosis requires integration of clinical history, morphologic changes, cytogenetics, and molecular analyses. Each of these components may change treatment strategies and ultimate outcome. Herein we describe the genetic and molecular features associated with the secondary AMLs and approaches and outcomes of conventional and emerging therapies.
CLINICAL CASE 1
A 55-year-old woman with a remote history of Burkitt lymphoma presented for routine annual examination and was found on complete blood count to have pancytopenia. White blood cell count was 900/µL, hemoglobin was 7.4 g/dL, and platelet count was 101 000/µL. Bone marrow examination revealed 28% blasts by morphology, and karyotype was complex 43,XX, del(5)(q11.2q33),-13,add(14) (p11.2),add(17)(p11.2),dic(18;19)(p11;2; q13.1),psu dic(22;1)(q13;p13)[16]/45,XX,del(5),der(13;14) (q10;q10), add(17),add(20)(q13.1)[2],46,XX[2]. Molecular studies revealed TP53 mutation at R248Q with 62% variant allele frequency (VAF). Performance status was excellent, and she had normal end- organ function. Her lymphoma history was notable for 30 years of ongoing remission after multiagent chemotherapy in her early 20 s. She had an unknown quantity of anthracycline exposure, and records of the therapy were unavailable. Echocardiogram at time of diagnosis revealed normal ejection fraction without regional wall motion abnormalities.
This patient has myeloid neoplasm, post–cytotoxic therapy, and her blasts >20% put her in the acute myeloid leukemia category. Cohorts of AML-pCT tend to contain more women than men as the most common primary malignancy is breast cancer followed by non-Hodgkin lymphoma.10 Despite the long latency from treatment to AML-pCT presentation, her complex karyotype and TP53 mutation are common features of AML-pCT. Multiple treatments are options in this case. Standard-of-care therapy with “7 + 3” (7 days of cytarabine and 3 days of anthracycline) would be the most common approach in this case, given a 33% to 55% overall response rate in secondary AML with intensive chemotherapy.1,11 However, the full molecular and karyotypic pattern would predict for a low overall response rate. Based on karyotypic changes alone, she also meets criteria for AML, myelodysplasia related,3,5 and as such, therapy with liposomal cytarabine and daunorubicin (CPX-351) could be considered.12 The overall response rates associated with secondary AML and TP53-mutant leukemia for both 7 + 3 and liposomal cytarabine/daunorubicin are summarized in Table 2. The long-term survival with either therapy is very limited and only found in patients who can proceed with allogeneic transplantation.13,14 The patient's history of high-grade lymphoma and extensive remote chemotherapy exposure would also complicate her tolerance of high-intensity chemotherapy. With limited knowledge about her anthracycline exposure, the choice of a regimen with anthracyclines becomes problematic even with normal ejection fraction and use of cardioprotection with dexrazoxane.
There were lower-intensity options offering reasonable remission rates as a bridge to curative intent transplant. Azacitidine and venetoclax (aza/ven) has been associated with complete response (CR) and complete remission with incomplete blood count recovery rate in AML with poor-risk cytogenetics and TP53 mutations of 41% in a recent review of older adults who were unfit for induction chemotherapy in phase 1b and phase 3 studies of aza/ven.15 Given no prospective comparative studies and a growing body of retrospective studies that show similar overall survival with venetoclax and hypomethylating agents compared with more traditional intensive chemotherapy approaches, a patient-centered discussion of putative risks and benefits is appropriate.16,17 This patient had a strong preference for outpatient-based therapy, and as such, we proceeded with this therapy. She achieved CR after 1 course and completed 5 cycles of aza/ven before proceeding to myeloablative allogeneic stem cell transplant 26 months ago with under 5% VAF TP53 mutation and normal karyotype at the time of transplant. She has ongoing remission and normal blood counts without active graft-vs-host disease.
Data on transplant in this secondary AML subset reflect outcomes predicted by pretransplant molecular and cytogenetic risks in de novo AML. Several studies support myeloablative conditioning over reduced intensity conditioning in improved relapse-free survival and equal or superior overall survival, although historically for TP53 mutation, the benefit has been less clear, especially in MDS.18 In addition, the presence of measurable residual disease (MRD), defined as detection of any of a subset of 13 commonly mutated genes on ultra-deep error-corrected sequencing of preconditioning blood, appears to confer the highest risk of relapse on patients who receive a nonmyeloablative transplant.19 It is unknown if MRD-negative patients may forgo myeloablative regimens.20 In this case, the achievement of remission allowed for transplantation to proceed. Despite the modest response rate published with aza/ven, the durability of these responses is often limited to less than 6 months, highlighting the need for more effective and durable therapy in this subset.15,21 Our current approach favors stem cell transplant when possible, but for intermediate- or adverse-risk patients who are ineligible or borderline candidates for transplant, novel therapies are needed.
CLINICAL CASE 2
A 52-year-old woman with a history of triple-negative breast cancer following adjuvant radiation therapy along with doxorubicin, cyclophosphamide, and docetaxel was found to have 40% blasts on routine blood work 12 months after completion of chemotherapy. Bone marrow biopsy specimen confirmed AML with 73% blasts, and chromosome analysis revealed 48,XX, +8,+13,t(16;16) (p13.1;q22)[20]. Molecular studies revealed a NRAS G13D mutation with a VAF of 38%. Her lifetime anthracycline dose was 240 mg/m2. She has no evidence of breast cancer at the time of diagnosis of AML.
Acute leukemia after exposure to ionizing radiation or chemotherapy rarely presents with otherwise favorable genetic markers such as fusions of RUNX1-RUNXT1, CBFB-MYH11, and PML-RARA in ~15% of cases.10 In the case of acute promyelocytic leukemia after chemotherapy, the outcomes appear as favorable as the outcomes in patients who had never been exposed to chemotherapy.22 On occasion, core binding factor AML is found in patients with prior chemotherapy exposure, and review of the outcomes of these patients suggests standard-of-care therapies are associated with shorter overall survival than de novo cases. That said, overall outcomes appear determined by additional adverse genetic features more so than by prior exposure to cytotoxic therapies.23 A retrospective collection of 69 patients with treatment-related core binding factor leukemia revealed patients with secondary cytogenetic abnormalities had longer overall survival than those without abnormalities. Presence of trisomy 8 and trisomy 22 and loss of X or Y chromosome were associated with improved survival in both the post–cytotoxic therapy and de novo subsets.23 In these situations, we advocate standard-of-care therapies with cytarabine-containing induction regimens plus gemtuzumab ozogamicin.24 This highlights a principle that the key feature of secondary AML is the specific genetic changes present rather than prior therapy or antecedent MDS or MPN.
In these cases, monitoring MRD via high-sensitivity polymerase chain reaction (PCR)–based testing of the blood should be performed to document an MRD-negative remission for therapy to continue with curative intent consolidation. RUNX1::RUNX1T1 or CBFB::MYH11 fusion genes corresponding to t(8;21) or inv(16/t(16;16) changes can be monitored by blood-level real-time quantitative PCR with high sensitivity (10–4 to 10–6), allowing potential early intervention.25 For these assays to be most reliable, assessment via molecular assays at the time of diagnosis is critical to establish the ability to detect fusions that occasionally can be missed if noncanonical gene fusions are present. Even in the secondary leukemia setting, patients with otherwise favorable leukemias can have reasonable outcomes with non-transplant-based therapy. Measurement of RUNX1-RUNX1T1 MRD via reverse transcription (RT)–PCR after course 1 of 7 + 3 with gemtuzumab and course 2 of high-dose cytarabine revealed no detectable evidence of disease, and the patient has completed 4 courses of consolidation without transplant in first CR.
CLINICAL CASE 3
A 69-year-old woman presented for shortness of breath and was found to have pancytopenia. She had no prior diagnosis of an antecedent MDS or MPN and no history of cytotoxic therapies. Bone marrow biopsy specimen revealed AML, myelodysplasia related. Blasts were enumerated at 68%, and dysplasia was greater than 50% in 2 cell lines. Karyotype was normal. Molecular testing revealed NPM1 (46% VAF), IDH1 R132H (48% VAF), SRSF2 (45% VAF), and FLT3-ITD (4.5% VAF). She is fit for induction chemotherapy.
AML-MR falls under a category of AML included in the secondary subset. This case highlights a common scenario where a patient presents without a known antecedent disorder and is still managed as having secondary AML. In this case, the founder mutation in the IDH1 gene at R132H likely predisposed the patient to the dysplastic features seen in this presentation. Therapeutic options include conventional therapy with 7 + 3 or CPX-351, which has been approved for patients with AML-MR.12 In this case, before knowing the results of the NPM1 and FLT3 testing, the patient initiated therapy with liposomal cytarabine/daunorubicin. Upon determination of the low-level FLT3-ITD mutation, midostaurin was added on days 8 to 21 based on improved overall survival with standard 7 + 3 induction therapy plus midostaurin in the RATIFY trial.26 She was found to have MRD-negative remission at the completion of induction via assessment of NPM1 with quantitative RT-PCR and proceeded to nonmyeloablative stem cell transplant given her age over 65 years.
This case highlights some complexity in the diagnostics and characterizations of AML with the evolution of novel therapies and evolved risk categorizations to include molecular changes. By the WHO 2016 classification system, the presence of dysplasia alone would have been sufficient for a diagnosis of AML-MRC, but by the 2022 classification system criteria, the presence of SRSF2 or other molecular or cytogenetic changes is necessary for AML-MR. By the European LeukemiaNet 2017 criteria, she would fall into favorable-risk disease.27 In younger patients, overall survival (OS) is similar in those who receive an allogenic stem cell transplant in first CR and those who do not. It is unclear if this finding can be extrapolated to older patients.28 While favorable via this method, the median OS in the phase 3 study of CPX-351 was only 9.7 months.12 As such, our standard practice is to consolidate remissions achieved in patients given CPX-351 who are transplant eligible with an allogeneic stem cell transplant. In the registrational study, the outcomes in patients who went to transplant after CPX-351 appear to be improved over those who received 7 + 3, although MRD assessments were not available.14 An interesting subanalysis of these OS data suggested lower transplant-related (nonrelapse) mortality in the CPX-351 arm.29 Specific to the NPM1 subset, quantitative RT-PCR monitoring of blood or marrow after induction can identify patients likely to relapse and those with intermediate-risk disease who would benefit from transplant, monitoring of blood once in remission can identify favorable-risk patients likely to relapse, and monitoring of blood or bone marrow before and after transplant can help prognosticate risk of relapse.30 In older adults in whom improvements from favorable-risk disease appear less,31 stem cell transplant still appears to be advantageous.32 This patient was fit for intensive therapy, but venetoclax and azacitidine would be any option for many other 69-year-old patients. We would have favored that approach over, say, ivosidenib and azacitidine given the higher response rate and faster time to response.
CLINICAL CASE 4
A 77-year-old man presented for evaluation of dizziness and shortness of breath and was found to have a white blood cell count of 7480/µL with 17% peripheral blood blasts, hemoglobin of 6.1 g/dL, and platelet count of 70 000/µL. Bone marrow examination revealed 48% blasts and chromosome analysis revealed a complex karyotype with evidence of monosomy 5, 7, and 17. Molecular studies revealed mutation in TP53 at R273 with a VAF of 80%. His performance status prior to this presentation was excellent for his age.
AML with complex karyotype and TP53 mutations appears to have the worst outcomes of the secondary AMLs. We estimate the likelihood of achieving remission with standard 7 + 3 and CPX-351 to be about 30% in these cases (Table 2). This patient may be considered ineligible for standard induction by age alone, but the biologic features of his disease make a stronger case for alternative therapies. In the phase 3 study of azacitidine and venetoclax, the remission rates were modest (55%), but the OS was short (6.5 months).21 There are several novel strategies under development specifically in the TP53 subset, including TP53 activating therapy in the form of eprenetapopt (APR-246). In early-phase studies, the reported response rates were high33 but, in subsequent randomized studies compared with azacitidine alone, failed to reach a primary end point of improvement in CR rates (Aprea press release 12/2020). The more recent strategies involve the use of immune target activation. There are several emerging immunotherapy strategies, including immune checkpoint blockade (such as Tim-3), bispecific antibody therapies (such as flotetuzumab, an investigational bispecific antibody-based molecule to CD3ε and CD123), and the most advanced candidate: monoclonal antibodies to CD47 (such as magrolimab). The expression of CD47 on myeloid cells acts to block immune response to malignant cells directly at the site of phagocytosis. Interestingly, CD47 messenger RNA expression varies across cytogenic and molecular subgroups, with lower expression in cases with t(8;21), a favorable-risk translocation, and higher expression in cases harboring FLT3-ITD mutations. High CD47 expression has been shown to be an independent prognostic factor for poor OS in AML patient cohorts.34 The monoclonal antibody magrolimab has been developed to block the CD47 signaling and in early-phase development shows promising activity against TP53 AML, with an overall response rate of 71% (48% CR and 19% CR with incomplete count recovery, 5% morphologic leukemia-free state) in 21 patients.35 The definitive randomized phase 3 studies in AML and MDS are enrolling. In addition, triplet therapy with venetoclax and a hypomethylating agent has been reported in abstract form, with a 64% CR rate in TP53-mutant disease. Fifty-five percent of these CRs were MRD negative by flow cytometry, suggesting deep remissions in a subset of the responders.36 In this patient, we elected to enroll him on a clinical trial examining the efficacy of a CD47 monoclonal antibody with the knowledge that transplant in this age group is generally not an option.
Conclusions
Secondary AML represents a heterogenous group of patients with features often overlapping de novo cases with similar genetics. Management and risks are often comparable to de novo disease with similar genetic features. Secondary AML encompasses a wide spectrum of molecular features. The historically poor outcomes in secondary AML reflect the higher proportion of negative features like poor risk cytogenetics and TP53 mutation. Furthermore, patients are frequently older with complex medical histories, limiting treatment options. The complete profiling of histopathology and genetic drivers is critical with the evolution of alternatives to standard 7 + 3 therapy. Patients with more favorable underlying cytogenetic and molecular features may do well with therapy, especially those who are candidates for allogeneic stem cell transplantation. Overwhelmingly, patients have disease with poor-risk features. Only rarely do patients have AML-pCT that is truly low risk. Maintenance therapy or transplantation is appropriate in most cases. Venetoclax-based regimens have expanded treatment options for frailer, older patients with stable comorbidities. Emerging treatments seek to open the therapeutic window by targeting specific genetic vulnerabilities or altering the microenvironment. Current treatment decisions require a careful integration of clinical history, genetic features of disease, and evaluation for allogeneic transplant.
Conflict-of-interest disclosure
Andrew Matthews: no competing financial interests to declare.
Keith W. Pratz: research funding from AbbVie, Agios, Daiichi Sankyo, and Millennium; advisory board member for AbbVie, Astellas, Boston BioMedical, BMS, Celgene, Novartis, Jazz Pharmaceuticals, and Servier.
Off-label drug use
Midostaurin is approved in combination with standard cytarabine and daunorubicin induction and cytarabine consolidation. Case discussion gives an example of a patient starting CPX-351 with addition of midostaurin after FLT3 mutation resulted.