Abstract
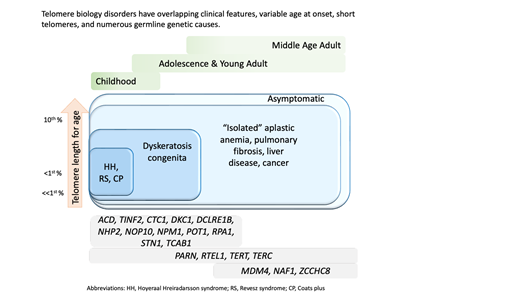
Numerous genetic discoveries and the advent of clinical telomere length testing have led to the recognition of a spectrum of telomere biology disorders (TBDs) beyond the classic dyskeratosis congenita (DC) triad of nail dysplasia, abnormal skin pigmentation, and oral leukoplakia occurring with pediatric bone marrow failure. Patients with DC/TBDs have very short telomeres for their age and are at high risk of bone marrow failure, cancer, pulmonary fibrosis (PF), pulmonary arteriovenous malformations, liver disease, stenosis of the urethra, esophagus, and/or lacrimal ducts, avascular necrosis of the hips and/or shoulders, and other medical problems. However, many patients with TBDs do not develop classic DC features; they may present in middle age and/or with just 1 feature, such as PF or aplastic anemia. TBD-associated clinical manifestations are progressive and attributed to aberrant telomere biology caused by the X-linked recessive, autosomal dominant, autosomal recessive, or de novo occurrence of pathogenic germline variants in at least 18 different genes. This review describes the genetics and clinical manifestations of TBDs and highlights areas in need of additional clinical and basic science research.
Learning Objectives
Identify the spectrum of clinical manifestations in telomere biology disorders, including variable age at onset and disease progression
Determine the appropriate diagnostic evaluation of patients with suspected telomere biology disorders, including appropriate genetic testing
Introduction
Dyskeratosis congenita (DC) and related telomere biology disorders (TBDs) comprise a heterogeneous group of illnesses caused by germline mutations resulting in aberrant telomere biology.1–5 Initially described in young males, DC was thought to occur primarily through X-linked recessive (XLR) inheritance. The discovery of mutations in dyskerin (DKC1) on the X chromosome resulting in reduced telomerase activity and short telomeres was the first connection between telomere biology and human disease.6,7 It is now well established that females are also affected, that the medical manifestations are broad, and that at least 18 different genes with all modes of inheritance—XLR, autosomal dominant (AD), and autosomal recessive—are etiologically connected.1–4
Dysplastic fingernails and toenails, oral leukoplakia, and lacy, reticular skin pigmentation define the mucocutaneous phenotype of DC.8 Patients with DC are at very high risk of bone marrow failure (BMF), cancer, PF, pulmonary arteriovenous malformations (PAVMs), liver disease, stenosis of the urethra, esophagus, or lacrimal ducts, avascular necrosis (AVN) of the hips and/or shoulders, and many other medical problems.1-3,9 One end of the clinical spectrum consists of very young children with DC, Hoyeraal-Hreidarsson (HH) syndrome, Revesz syndrome (RS), or Coats plus. The other end of the spectrum includes middle-aged or older adults with one feature, such as PF.10
How does one reconcile the clinical and genetic heterogeneity of such a complex hereditary disorder? These illnesses have had many names (Zinsser-Engman-Cole syndrome, DC, telomeropathies, short telomere syndromes, impaired telomere maintenance spectrum disorders, and TBDs), but the common thread is their etiology—germline mutations resulting in abnormal telomere biology—hence the designation TBD.2,11
Case presentations
These individuals were participants in the National Institutes of Health Institutional Review Board–approved Inherited Bone Marrow Failure Syndromes study (ClinicalTrials.gov identifier NCT00027274, https://marrowfailure.cancer.gov).12 The patient or their legal guardian(s) signed informed consent.
CLINICAL CASE 1
A three-year-old previously healthy girl presented to her pediatrician with petechiae and anemia after a viral illness. Persistent cytopenias over the next 6 months led to a diagnosis of acquired aplastic anemia. She was treated with antithymocyte globulin and cyclosporin without an initial response and failed another trial 6 months later. By 6 years of age, she was dependent on red blood cell transfusions, occasionally required platelet transfusions, and had leukopenia. Trials of granulocyte colony-stimulating factor, erythropoietin, and androgens did not improve her cytopenias. The patient underwent a successful matched unrelated nonmyeloablative hematopoietic cell transplantation (HCT) at 9 years of age. After HCT, abnormal fingernails and toenails, oral leukoplakia, and excessive tearing were reported (Figure 1A). Evaluations for graft-versus-host disease were negative.
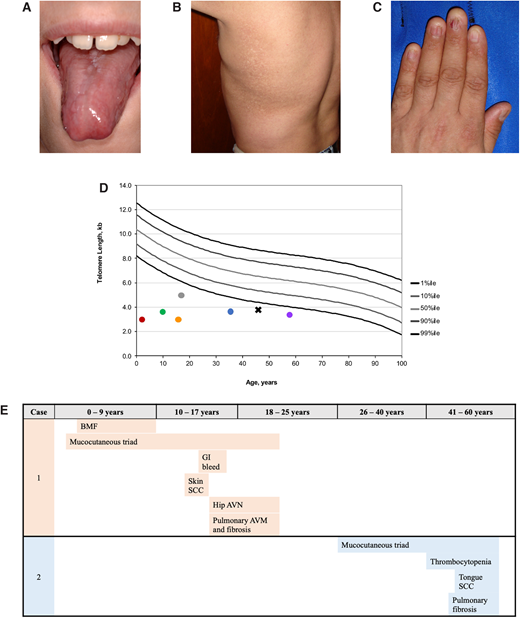
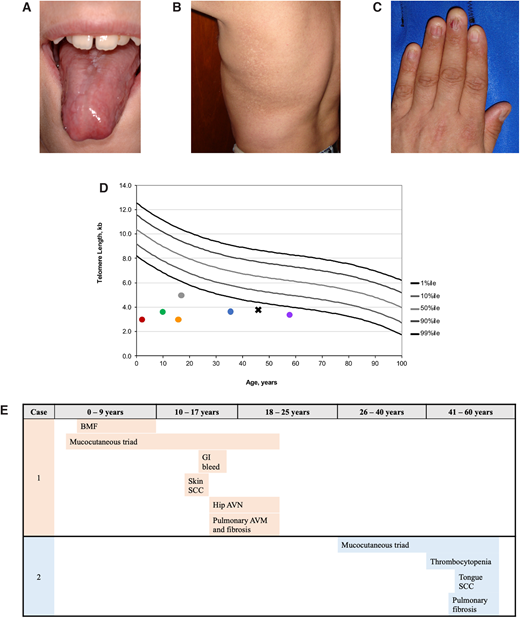
Examples of clinical manifestations of telomere biology disorders. (A) Tongue leukoplakia in Clinical Case 1; (B) hyper- and hypopigmentation on the trunk in Clinical Case 2; (C) nail dysplasia in Clinical Case 2; (D) examples of lymphocyte telomere lengths measured by flow cytometry with in situ hybridization from NCT00027274 study participants. Circle color codes by sex and causative gene: red, male with heterozygous TINF2; green, male with DKC1; gray, male with DKC1; orange, female with autosomal recessive RTEL1; blue, male with heterozygous TERT; purple, female with heterozygous TERC. Clinical Case 2 is designated with a black X; (E) TBD phenotype progression in Clinical Cases 1 and 2 by age group with age at diagnosis noted.
AVM, arteriovenous malformation; SCC, squamous cell carcinoma.
Examples of clinical manifestations of telomere biology disorders. (A) Tongue leukoplakia in Clinical Case 1; (B) hyper- and hypopigmentation on the trunk in Clinical Case 2; (C) nail dysplasia in Clinical Case 2; (D) examples of lymphocyte telomere lengths measured by flow cytometry with in situ hybridization from NCT00027274 study participants. Circle color codes by sex and causative gene: red, male with heterozygous TINF2; green, male with DKC1; gray, male with DKC1; orange, female with autosomal recessive RTEL1; blue, male with heterozygous TERT; purple, female with heterozygous TERC. Clinical Case 2 is designated with a black X; (E) TBD phenotype progression in Clinical Cases 1 and 2 by age group with age at diagnosis noted.
AVM, arteriovenous malformation; SCC, squamous cell carcinoma.
CLINICAL CASE 2
A previously healthy 43-year-old man was found to have moderate thrombocytopenia at a routine health care visit. It was presumed to be idiopathic thrombocytopenia, and his blood counts were followed, but no treatment was initiated. At 45 years of age, he saw a dermatologist for nail and skin pigmentation abnormalities, and this physician suggested DC (Figure 1B-C).
Diagnosing TBDs
Telomeres consist of (TTAGGG)n repeats and a protein complex at the chromosome ends essential for chromosomal stability.13,14 Their nucleotide repeats shorten with each cell division due to the inability of DNA polymerase to fully replicate the 3′ ends of DNA. When telomeres reach a critical length, cells undergo apoptosis or enter senescence. Human telomere lengths are highly variable, decline with age, and are estimated to range from 2 to 14 kilobases depending on cell type, age, genetic factors, ancestry, and measurement method.15 Patients with TBDs have very short telomeres for their age due to germline mutations in key telomere biology genes.
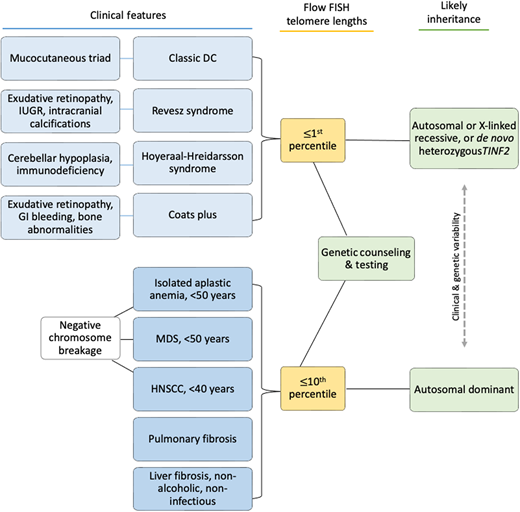
Clinical features diagnostic of classic DC include (1) all three mucocutaneous triad features (nail dysplasia, lacy skin pigmentation, and oral leukoplakia) or (2) any 1 feature of the triad in combination with BMF and 2 other physical findings consistent with DC.3,4,16 Patients with cerebellar hypoplasia and immunodeficiency in addition to DC features may be diagnosed with HH. RS syndrome is often differentiated from DC by the addition of bilateral exudative retinopathy. Patients with Coats plus primarily have cerebroretinal microangiopathy with calcifications and cysts, exudative retinopathy, abnormal bone healing, and gastrointestinal (GI) bleeding due to vascular ectasias.17
Flow cytometry with fluorescent in situ hybridization (flow FISH) of leukocytes is the current diagnostic test for TBDs (Figure 1D). Other telomere length measurement methods are used on a research basis but are not yet validated for clinical diagnostics, including terminal restriction fragment (TRF) measurement by Southern blot,18 quantitative polymerase chain reaction (qPCR), and single telomere length assays.19,20
Flow FISH lymphocyte telomere lengths less than the first percentile for age are highly sensitive and specific for diagnosing patients with TBDs.21,22 In fact, total lymphocyte flow FISH telomere lengths less than the first percentile for age have a sensitivity and specificity of 97% and 91%, respectively, for differentiating patients with DC from their unaffected relatives. Granulocyte telomere lengths less than the first percentile are similarly sensitive (97%) but not specific (82%) and thus do not usually aid in diagnosis. The measurement of telomere lengths in lymphocyte subsets can sometimes aid in the diagnosis of borderline cases by increasing the sensitivity and specificity to 98% and 94%, respectively, if telomeres are below the first percentile for age in more than or equal to 3 out of 4 lymphocyte subsets (CD45RA+/CD20− naive T cells, CD45− memory T cells, CD20+ B cells, and/or CD57+ natural killer/natural killer T cells). Importantly, telomeres at or below the first percentile rarely occur in patients with other inherited BMF syndromes, such as Fanconi anemia, Diamond-Blackfan anemia, or Shwachman-Diamond syndrome.21,23,24
Adults presenting with a new TBD diagnosis, such as aplastic anemia, myelodysplastic syndrome (MDS), PF, or noninfectious, nonalcoholic liver fibrosis may have telomeres in the 1st to 10th percentile for age.9,25 This finding is also consistent with genotype-phenotype analyses finding fewer clinical complications in adults with heterozygous pathogenic variants.4,21 In these instances, identifying the genetic etiology is important in determining treatment.3
Patients may develop TBD features at any age and in any order (Figure 1E).4,8,12,26–28 Children may develop BMF prior to the mucocutaneous triad. They may present with immunodeficiency and esophageal stenosis prior to other complications. An adult with abnormal nails and mild thrombocytopenia may not be considered for TBD evaluation until their parent develops PF. All patients with new-onset BMF should be assessed for Fanconi anemia by chromosome breakage analysis, leukocyte flow FISH telomere length testing, and a germline genetic panel for BMF susceptibility genes (Figure 2).16
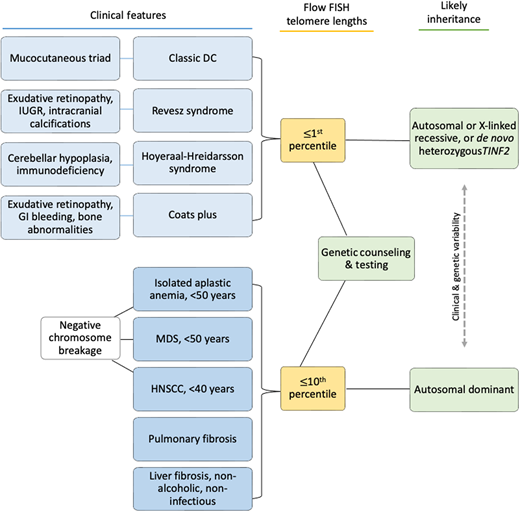
Clinical features underlying TBDs and possible diagnostic evaluation results. Telomere length testing should be considered for all patients with the indications shown. All patients with BMF or head/neck squamous cell carcinoma (HNSCC) should also have chromosome breakage testing of blood (and skin, if indicated) to rule out Fanconi anemia. Family history may be helpful if it is present, but many patients do not have affected relatives due to variable disease penetrance, expressivity, and/or genetic anticipation.
HNSCC, head and neck squamous cell carcinoma; IUGR, intrauterine growth restriction.
Clinical features underlying TBDs and possible diagnostic evaluation results. Telomere length testing should be considered for all patients with the indications shown. All patients with BMF or head/neck squamous cell carcinoma (HNSCC) should also have chromosome breakage testing of blood (and skin, if indicated) to rule out Fanconi anemia. Family history may be helpful if it is present, but many patients do not have affected relatives due to variable disease penetrance, expressivity, and/or genetic anticipation.
HNSCC, head and neck squamous cell carcinoma; IUGR, intrauterine growth restriction.
CLINICAL CASE 1 (Continued)
The patient was evaluated at 10 years of age, after HCT. The presence of nail dysplasia, abnormal skin pigmentation, and oral leukoplakia was consistent with DC, but it was not possible to confirm the diagnosis by flow FISH telomere length measurement because her lymphocytes were from the HCT donor. Relative telomere length was determined by qPCR on a research basis using DNA from the patient's primary fibroblast cell culture and was consistent with DC. However, it is important to note that while qPCR is highly specific for DC/TBDs, it is not sensitive enough for diagnostics and not currently used in the clinic.29
CLINICAL CASE 2 (Continued)
The patient's evaluation at 45 years of age showed skin and nail manifestations consistent with DC as well as flow FISH telomere lengths less than the first percentile for age (Figure 1D).
The genetic etiology of TBDs
It is important to determine whether any of the TBD-associated clinical features (Table 1) are present in the patient's relatives. Family history can guide genetic diagnosis of TBDs in many cases, but variable expressivity, incomplete penetrance, and genetic anticipation can complicate the assessment.5,30 Telomere length testing of family members may also be helpful in determining inheritance patterns.
Genetic education and counseling are essential for all individuals undergoing a TBD evaluation because the results may have far-reaching implications related to clinical prognostication and family planning.31 Individuals being tested, or their guardians, need to understand the complex clinical spectrum, the modes of inheritance, and the implications of genetic testing for the entire family.
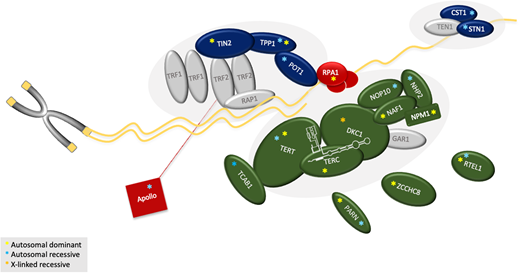
The genes currently associated with TBDs, their modes of inheritance, and their function are shown in Table 2 and Figure 3. Most inherited BMF or familial PF genetic testing panels contain many, but not consistently all, of the known DC/TBD-associated genes (ACD, CTC1, DKC1, NAF1, NHP2, NOP10, PARN, POT1, RTEL1, STN1, TERC, TERT, TINF2, WRAP53, ZCCHC8).32 Other genes to consider, but not routinely, on panels are the recently discovered RPA1 and DCLRE1B, as well as MDM4, a DC-like gene reported in 1 family.33–35 If gene panel testing is inconclusive, exome sequencing on a clinical or research basis should be considered. Genetic testing may still be inconclusive because about 20% of patients do not have an identifiable genetic cause of their disease.1,3,4,12
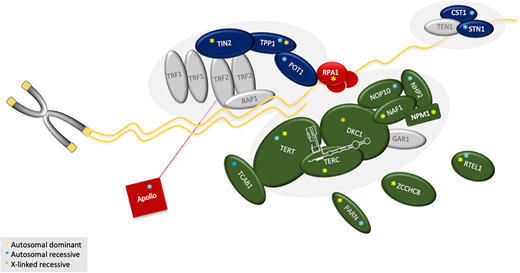
Schematic of the telomere and functions of the proteins affected in TBDs. Protein function and the reported consequences of mutations in the respective gene are summarized in Table 2. Components are grouped approximately based on their function. Shown in green: the components of the telomerase enzyme complex (DKC1, TERC, TERT, NAF1, NOP10, NHP2), telomerase or hTR regulators (TCAB1, PARN, and ZCCHC8), and regulator of telomere elongation helicase 1 (RTEL1). Dark blue: the shelterin (TPP1, TIN2, and POT1) and the CST (CTC1 and STN1) complexes. Red: the proteins primarily involved in DNA repair. Red line: Apollo interacts with TRF1. NPM1 is involved in ribosomal RNA maturation and interacts with NOP10 and NHP2. MDM4 is not shown because it does not directly act at the telomere. Gray symbols: known key telomere biology proteins not yet attributed to human disease (GAR1 in telomerase complex; TRF1, TRF2, RAP1 in shelterin; TEN1 in CST). Yellow asterisk: autosomal dominant; light-blue asterisk: autosomal recessive; orange asterisk: X-linked recessive. Complete protein name (gene name): Apollo (DCLRE1B); 1, CTC1, conserved telomere maintenance component 1 (CTC1); DKC1, dyskerin (DKC1); MDM4, MDM4 regulator or p53 (MDM4); NAF1, nuclear assembly factor 1 ribonucleoprotein (NAF1); NHP2, NOLA2 nucleolar protein family A, member 2 (NHP2); NOP10, nuclear protein family A, member 3 (NOP10); NPM1, nucleophosmin 1 (NPM1); PARN, poly (A)-specific ribonuclease (PARN); POT1, protection of telomeres 1 (POT1); RPA1, replication protein A1 (RPA1); RTEL1, regulator of telomere elongation helicase 1 (RTEL1); STN1, CST complex subunit (STN1); TCAB1, telomere Cajal body–associated protein 1 (WRAP53); TERC, hTR, human telomerase RNA component (TERC); TERT, human telomerase reverse transcriptase (TERT); TIN2, TRF1-interacting nuclear factor 2 (TINF2); TPP1: telomere protection protein 1 (ACD); ZCCHC8: Zinc finger CCHC domain-containing protein 8 (ZCCHC8).
Figure adapted with permission from Niewisch and Savage.1
Schematic of the telomere and functions of the proteins affected in TBDs. Protein function and the reported consequences of mutations in the respective gene are summarized in Table 2. Components are grouped approximately based on their function. Shown in green: the components of the telomerase enzyme complex (DKC1, TERC, TERT, NAF1, NOP10, NHP2), telomerase or hTR regulators (TCAB1, PARN, and ZCCHC8), and regulator of telomere elongation helicase 1 (RTEL1). Dark blue: the shelterin (TPP1, TIN2, and POT1) and the CST (CTC1 and STN1) complexes. Red: the proteins primarily involved in DNA repair. Red line: Apollo interacts with TRF1. NPM1 is involved in ribosomal RNA maturation and interacts with NOP10 and NHP2. MDM4 is not shown because it does not directly act at the telomere. Gray symbols: known key telomere biology proteins not yet attributed to human disease (GAR1 in telomerase complex; TRF1, TRF2, RAP1 in shelterin; TEN1 in CST). Yellow asterisk: autosomal dominant; light-blue asterisk: autosomal recessive; orange asterisk: X-linked recessive. Complete protein name (gene name): Apollo (DCLRE1B); 1, CTC1, conserved telomere maintenance component 1 (CTC1); DKC1, dyskerin (DKC1); MDM4, MDM4 regulator or p53 (MDM4); NAF1, nuclear assembly factor 1 ribonucleoprotein (NAF1); NHP2, NOLA2 nucleolar protein family A, member 2 (NHP2); NOP10, nuclear protein family A, member 3 (NOP10); NPM1, nucleophosmin 1 (NPM1); PARN, poly (A)-specific ribonuclease (PARN); POT1, protection of telomeres 1 (POT1); RPA1, replication protein A1 (RPA1); RTEL1, regulator of telomere elongation helicase 1 (RTEL1); STN1, CST complex subunit (STN1); TCAB1, telomere Cajal body–associated protein 1 (WRAP53); TERC, hTR, human telomerase RNA component (TERC); TERT, human telomerase reverse transcriptase (TERT); TIN2, TRF1-interacting nuclear factor 2 (TINF2); TPP1: telomere protection protein 1 (ACD); ZCCHC8: Zinc finger CCHC domain-containing protein 8 (ZCCHC8).
Figure adapted with permission from Niewisch and Savage.1
Determining whether a specific genetic variant is pathogenic, a variant of uncertain significance, or benign is a complex process requiring in-depth understanding of the protein function, gene expression, bioinformatic predictions, and frequency in healthy and affected populations. All providers are strongly encouraged to follow the American College of Medical Genetics and Genomics and the Association for Molecular Pathology frequently updated variant curation recommendations.36
CLINICAL CASE 1 (Continued)
The patient's sibling, parents, and grandparents were healthy, with no reported features of TBDs. The telomere lengths of her sibling and parents were greater than the 10th percentile for their ages. Genetic testing of DNA from skin fibroblast cultures identified a TINF2 pathogenic variant in the patient but not in her first-degree relatives. TINF2 mutations are frequently de novo in TBDs, although some families with AD inheritance have been reported.5,37
CLINICAL CASE 2 (Continued)
The patient's family history was notable for his mother, who died due to nonalcoholic, noninfectious liver cirrhosis. She had also received many red blood cell transfusions and had abnormal nails and alopecia. The patient's maternal grandmother also had alopecia and abnormal nails. Although the patient's family history is consistent with AD inheritance, a pathogenic germline variant (ie, mutation) was identified in DKC1, the XLR gene. Genetic testing of his mother and maternal grandmother was not possible, but his sister was heterozygous for the same DKC1 mutation. This finding brings up two key points: (1) TBDs can manifest in adults, even if due to genes usually associated with childhood onset, and (2) skewed X-chromosome inactivation in female carriers of DKC1 mutations should be considered in their differential diagnosis.38,39
Clinical manifestations and management
It is important for clinicians, patients, and their family members to be proactive in addressing the high risk of the multiple progressive and life-threatening complications associated with TBDs. The clinical complications and care recommendations are listed in Table 1. The reader is also referred to the second edition of Telomere Biology Disorders: Diagnosis and Management Guidelines, published in 2022 by Team Telomere, the DC/TBD patient support and advocacy group.40 The new Guidelines includes the latest expert recommendations and can be freely obtained at https://teamtelomere.org/diagnosis-management-guidelines/. Additional resources for clinicians, patients, and families are available at Team Telomere (www.teamtelomere.org) and/or DC Action (http://dcaction.org).
CLINICAL CASE 1 (Continued)
In retrospect, the diagnosis of classic DC was likely present in the patient prior to HCT (Figure 1E). Abnormal nails were present when she was a toddler, but neither her physicians nor parents thought they were significant at the time. Additionally, she developed BMF when telomere length testing was not available and DC was still thought to be an XLR disease. Her nail dystrophy, abnormal skin pigmentation, and oral leukoplakia progressed over the course of her life. Skin squamous cell carcinoma was resected at 13 years of age. At about age 14, she developed GI bleeding that was eventually attributed to small vascular ectasias in the stomach and small intestine. Hip pain developed between 13 and 14 years of age, and x-ray showed AVN of the femoral heads. Hip replacements were considered but not performed because of her pulmonary status. She developed shortness of breath with exertion at age 15, which gradually progressed. Extensive evaluation identified small PAVMs and PF. Her symptoms progressed and, unfortunately, she died before a lung transplant could be performed.
CLINICAL CASE 2 (Continued)
This patient's clinical course is illustrative of adult-onset TBDs (Figure 1E). His thrombocytopenia was initially moderate, but his platelet counts gradually declined. He also developed progressive anemia and was started on oral danazol at age 49. His blood counts improved, and he did not require transfusions. Baseline pulmonary function tests at age 45 showed a restrictive pattern and mild diffusion defect, and mild to moderate PF was present on high-resolution computed tomography scan. Unfortunately, he developed squamous cell carcinoma of the tongue at 53 years of age, requiring extensive surgery. Radiation therapy was administered but complicated by severe mucositis and poor wound healing. He died less than a year after his cancer diagnosis.
Genotype-phenotype associations
TBD's clinical manifestations and a patient's telomere length often vary by gene and mode of inheritance. Patients with heterozygous mutations (except for TINF2) tend to present later in life, with fewer medical problems, whereas those with XLR or autosomal recessive disease are usually diagnosed in childhood. The clinical, genetic, and telomere length heterogeneity, in addition to its relative rarity, makes it difficult to fully assess genotype-phenotype associations (Figure 1D).
Our study of 231 individuals grouped patients into 3 categories (1) AD dominant non-TINF2 (AD-nonTINF2), (2) autosomal recessive and XLR (AR/XLR), and (3) TINF2.4 TINF2 was considered separately because it frequently occurs de novo. Patients with AD-nonTINF2 had better overall survival, at 64.9 years (95% CI, 59.8-67.6), than those with AR/XLR (31.8 years; 95% CI, 24.2-37.4) or TINF2 (27.8 years; 95% CI, 19.1-39.1). These associations remained significant after accounting for telomere length (AR/XLR/TINF2 vs AD-nonTINF2: hazard ratio [HR], 4.8; P < 0.01). Those with AR/XLR or TINF2 TBDs had earlier onset of BMF, liver disease, and GI telangiectasias compared with the AD-nonTINF2 group. The age-adjusted cancer risk was high in all groups but highest in those with AR/XLR TBDs. PF was more likely in the AD-nonTINF2 patients.
Data are lacking on the acquisition of somatic mutations in patients with TBDs. There are a few reports of hematopoietic somatic reversion occurring in patients with TBDs due to DKC1, RPA1, TERC, TERT, or TINF2.33,41 A 2021 study performed ultradeep sequencing of 16 telomere biology genes (plus TP53) and found blood-derived DNA somatic mutations in 16 of 56 (29%) patients compared with none of the healthy controls (n = 28).42 Out of 16 patients with TBDs and MDS/acute myeloid leukemia (AML), 6 had somatic mutations. Seven candidate genes (TP53, the TERT promoter, POT1, TERF2IP, RBM7, SKIV2L2, and DIS3) had 36 clonal somatic mutations. Depending on the timing in the patient's clinical course and the specific hematopoietic stem cell acquiring a somatic mutation, patients with cytopenias may have gradual improvement of blood counts, or patients may have normal hematopoiesis despite other TBD-related complications. Somatic reversion can complicate genetic diagnosis, and a skin biopsy may be required.
Large collaborative studies are required to obtain the statistical power required to determine whether the acquisition of somatic mutations in TBDs could be used in clinical risk stratification.
What is next?
The last 2 decades have led to great progress in understanding the causes and complications of TBDs. The growing recognition of the role of aberrant germline telomere biology in human disease is leading to important advances in understanding the full scope of TBDs and the role of telomere biology in the general population. The prevalence of TBDs has been roughly estimated at 1 case per million,3 with approximately 900 to 1000 cases published to date.1 This is likely an underestimate, and additional genetic and phenotyping studies are required to fully quantify the extent to which TBDs exist.
This improved understanding of TBD genetic etiology calls for more studies of the specific cellular and molecular consequences of germline mutations affecting telomere biology. As more rare germline genetic variants are discovered in patients with known or suspected TBD, proper variant classification is required to aid in diagnosis, guide therapy and counseling, and inform basic science studies. Formation of an American College of Medical Genetics and Genomics/Association for Molecular Pathology Variant Curation Expert Panel through the Clinical Genome Resource is underway for TBD-associated genes.43
TBD-specific clinical trials are essential to improving clinical outcomes. There are only 4 therapeutic trials listed as recruiting participants as of May 2022 (https://clinicaltrials.gov)—1 on HCT (ClinicalTrials.gov identifier NCT01659606), 2 on danazol and telomere length (NCT03312400 and NCT04638517), and 1 gene therapy trial (NCT04211714). Another study is focused on understanding the psychosocial challenges of living with TBDs (NCT04959188).
Patients with TBDs should be included in clinical trials of PF, liver disease, PAVMs, and cancer. Outcomes should be analyzed according to whether a patient has a TBD and, if statistical power permits, by gene or mode of inheritance. TBD clinical and genetic heterogeneity requires multispecialty clinical care, multi-institutional scientific collaboration, and input from the patient community. The Clinical Care Consortium of Telomere-Associated Ailments is an international consortium of clinicians and scientists who agree to share deidentified data to learn more about TBD manifestations, with the goal of improving outcomes, and welcome the participation of new investigators.44
Collaboration between the research, clinical, and patient communities ensures many future steps aimed at improving the lives of all those affected by TBDs.
Acknowledgments
I am especially grateful to all the patients and families with DC/TBDs for their invaluable contributions to science and medicine. Thank you to Dr Neelam Giri, National Cancer Institute (NCI), and Dr Marena Niewisch, NCI, and University of Hannover, who provided much appreciated feedback on this manuscript.
This work was supported by the Intramural Research Program, Division of Cancer Epidemiology and Genetics, NCI.
Conflict-of-interest disclosure
Sharon A. Savage: no competing financial interests to declare.
Off-label drug use
Sharon A. Savage: nothing to disclose.