
Abstract
This is a comprehensive overview on the most recent developments in diagnosis and treatment of acute lymphoblastic leukemia (ALL).
Dr. Dieter Hoelzer and colleagues give an overview of current chemotherapy approaches, prognostic factors, risk stratification, and new treatment options such as tyrosine kinase inhibitors and monoclonal antibodies. Furthermore the role of minimal residual disease (MRD) for individual treatment decisions in prospective clinical studies in adult ALL is reviewed.
Drs. Ching-Hon Pui and Mary Relling discuss late treatment sequelae in childhood ALL. The relation between the risk of second cancer and treatment schedule, pharmacogenetics, and gene expression profile studies is described. Also pathogenesis, risk factors, and management of other complications such as endocrinopathy, bone demineralization, obesity, and avascular necrosis of bone is reviewed.
Dr. Fred Appelbaum addresses long-term results, late sequelae and quality of life in ALL patients after stem cell transplantation. New options for reduction of relapse risk, e.g., by intensified conditioning regimens or donor lymphocyte infusions, for reduction of mortality and new approaches such as nonmyeloablative transplantation in ALL are discussed.
Drs. Jacques van Dongen and Tomasz Szczepanski demonstrate the prognostic value of MRD detection via flow cytometry or PCR analysis in childhood ALL. They discuss the relation between MRD results and type of treatment protocol, timing of the follow-up samples, and the applied technique and underline the importance of standardization and quality control. They also review MRD-based risk group definition and clinical consequences.
I. Clinical Management of Adult ALL
Dieter Hoelzer, MD PhD,*
University Hospital, Medizinische Klinik III, Theodor Stern Kai 7, 60590 Frankfurt, Germany
In adult acute lymphoblastic leukemia (ALL) complete remission (CR) rates of 80-85% and leukemia-free survival (LFS) rates of 30-40% can be achieved (Table 1 ). Intensified consolidation, particularly with high-dose methotrexate (HDM) and high-dose cytarabine (HDAC), could be one reason for the improved outcome in recent studies. In addition more experienced participating centers or more rigorous stem cell transplantation (SCT) could contribute to better outcome. ALL subtypes show considerable differences in terms of clinical features, laboratory values, treatment response, relapse sites, and relapse kinetics. There has been substantial progress in some subtypes of ALL such as thymic or mature B-ALL with > 50% survival, whereas only limited improvement was achieved in the large group of B-precursor ALL and none in the Ph/BCR-ABL positive ALL, with LFS rates of 10-20% only.
Management and New Treatment Options
In adult ALL it seems, however, that with intensification of chemotherapy and more SCT the chance for further improvement is limited. This is particularly true for the high proportion of elderly ALL patients not covered in many trials. Thus it is fortunate that in the last few years promising new treatment modalities have come up.13 These new options in the treatment of adult ALL include molecular targeting with kinase inhibitors that selectively inhibit molecular aberrations involved in pathogenesis. Antibody therapy offers an additional targeted treatment approach without typical chemotherapy-associated toxicities. The extension of SCT should include new approaches that rely on graft-versus-leukemia effects such as non-myeloablative transplantation (NMSCT) and the application of donor leukocyte infusions. The evaluation of minimal residual disease (MRD) is an important step for individual evaluation of treatment response after achievement of CR. Finally microarray technology may be used for identification of new prognostic factors and targets for molecular therapeutics (Table 2 ).
Chemotherapy and Stem Cell Transplantation
Results obtained with chemotherapy, partly including SCT, in adult ALL are evident from Table 1. The table lists large multicenter studies with more than 500 patients, which probably provide the most realistic impression of outcome of adult ALL, but also more recent smaller studies with more than 100 patients, which demonstrate favorable results with intensive treatment approaches.
Induction therapy
Usually induction chemotherapy includes prednisone, vincristine, anthracyclines (mostly daunorubicin) and also L-asparaginase. Additional drugs such as cyclophosphamide, cytarabine (either conventional or high dose), mercaptopurine and others are added in many protocols sometimes named as early intensification. Several new approaches are currently being explored in adult ALL to improve CR rates and thereby remission quality.
Dexamethasone
Dexamethasone is used instead of prednisone since it may exert higher antileukemic activity toward systemic disease but also higher drug levels in the cerebrospinal fluid. Extensive use of dexamethasone may, however, be associated with an increased risk of septicemias and fungal infections,14 which may be circumvented if application time and dose are reduced.
Cyclophosphamide
The role of cyclophosphamide (C), generally administered at the beginning of induction therapy, has been evaluated in several studies. A randomized study by the Italian GIMEMA group comparing a 3-drug induction with and without C did not show a difference in terms of CR rate (81% vs 82%).6 However, in several non-randomized trials high CR rates (85-91%) were achieved with regimens including C pretreatment,8,10 particularly in adult T-ALL.15
Anthracycline
Anthracycline dose intensity and schedule may play an important role in induction therapy of ALL.16 Formerly daunorubicin was mostly administered at a weekly schedule, but recently many trials include dose intensification with doses of 30 to 60 mg/m² at a 2-3 day schedule.8,9,17 A particularly high CR rate (93%) was reported for intensive anthracycline therapy (270 mg/m² for 3 days)17 from a monocenter study, but was not seen in a larger multicenter trial.18 Intensive anthracycline therapy may be associated with a higher induction mortality. Therefore, intensive supportive care and probably the use of growth factors are recommended with these types of protocols.
L-asparaginase
For L-asparaginase (A), also a common part of induction therapy, three different preparations with significantly different half-lifes are available: Native E. coli A (1.2 days), Erwinia A (0.65 days), and PEG-L-A (5.7 days).19 In order to reach equal efficacy the application schedule has to be adapted, which is generally daily for Erwinia, every other day for E. coli, and 1-2 times weekly for PEG-A. The importance of A pharmacokinetics is illustrated by a randomized trial in childhood ALL where significantly lower survival rates were achieved with Erwinia compared to E. coli A, both given at the same schedule.20 A randomized trial comparing PEG-A and E. coli A in childhood ALL showed a higher earlier response rate for the latter but no difference in long-term outcome.21
High-dose cytarabine
High-dose cytarabine (HDAC) in induction therapy has been used in several trials in order to achieve higher antileukemic activity and in addition prophylaxis of central nervous system (CNS) relapse without cranial irradiation. Up-front application before conventional chemotherapy yielded higher CR rates than application afterward, which was in part related to a higher induction mortality. Furthermore any type of induction therapy with HDAC may lead to an increased incidence of severe neutropenias after subsequent chemotherapy cycles.22
Growth factors
With enhanced dose intensity of induction therapy the prophylactic use of growth factors has an increasingly important role in treatment of adult ALL. Several studies have shown that G-CSF can be administrated parallel to induction therapy and can significantly reduce the duration of neutropenias.23,8 In a placebo-controlled study there was also a higher CR rate with G-CSF (90% vs 81%) due to a lower early mortality (4% vs 11%).8 Thus with G-CSF support probably a higher dose intensity and better tolerability of chemotherapy may be achieved. It remains, however, open whether this translates to an improved LFS or overall survival.
Postinduction therapy
Postinduction therapy mainly consists of intensive rotational consolidation therapy, high-dose chemotherapy cycles, and SCT. There is generally a superior outcome from trials implementing intensive multidrug consolidation therapy (median: 27-36%) compared to those without consolidation (median: 25%).13 This was also confirmed in single randomized studies3 or historical comparisons1,10 but not in others.6,7 Scheduling of intensification (earlier superior to late) may play an important role. High-dose chemotherapy, mainly HDAC or HDM, has been used to overcome drug resistance and to achieve therapeutic drug levels in the cerebrospinal fluid. The overall impression is that the inclusion of HDAC, HDM or both might be beneficial particularly if included in regimens with intensive rotational conventional dose chemotherapy with LFS rates > 40%, but all in small series.13
Stem cell transplantation
Stem cell transplantation from bone marrow grafts and to an increasing extent transplantation of peripheral blood stem cells (PBSCT) is an essential part of consolidation treatment in adult ALL. The question is whether all patients in first CR with suitable sibling donor should receive allogeneic SCT or whether it should be reserved for patients with high risk features. Furthermore the value of matched unrelated SCT, which may be associated with a lower relapse rate, autologous SCT, and new approaches such as nonmyeloablative transplantation, new conditioning regimens, e.g., with radiolabeled antibodies and donor leukocyte infusions, needs to be determined. Several recent or ongoing studies include early SCT (mainly from matched related donors) but so far it has not been demonstrated that this approach has an impact on overall outcome (see also Section III).
Prognostic Factors
Although prognostic factors for response and LFS depend on the treatment regimen, they appear similar in large adult ALL trials, indicating that treatment approaches are not entirely different. In adult ALL the most important prognostic factors such as age, white blood cell count (WBC), immunophenotype, cytogenetics, and molecular genetics are determined at diagnosis. Clinical parameters such as CNS involvement or mediastinal tumors are of less or no significant prognostic value. On the other hand response to treatment is significantly correlated with outcome. This includes the time to achievement of CR, mostly analyzed after 2-4 weeks and level and course of MRD during induction and consolidation (Table 3 ).
Immunologic subtypes
The immunologic subtypes of ALL show considerable differences in terms of presentation, clinical course, and relapse risk. Thus the formerly unfavorable subgroup pro B-ALL, which is characterized by a high proportion of t(4;11)/ALL1-AF4 positive ALL (70%) and high WBC at presentation (> 100.000/μL in 26%), now reaches LFS rates above 50% with regimens including HDAC and particularly allogeneic SCT.26 Common(c)/pre-B-ALL shows a high incidence of Ph/BCR-ABL positive ALL (40-50%). It can be subdivided into a standard and a high-risk group (Table 3) with significantly different outcome. In c/pre-B-ALL experience from childhood ALL shows that treatment with HDM may be particularly effective. It remains to be demonstrated, however, whether similarly intensive consolidation is applicable in adult ALL. Furthermore leukemic blasts in c/pre-B-ALL express several antigens (CD19, CD20, CD22) that could be targets for antibody therapy. Since no prognostic factors are known in standard risk c-/pre-B-ALL the evaluation of MRD may help to identify patients with high risk of relapse who could benefit from intensification, e.g., by SCT.
Mature B-ALL is generally treated according to a different concept, with short intensive cycles without maintenance therapy. It is characterized by a high rate of organ (32%) and CNS (12%) involvement. The regimens include fractionated C or ifosfamide, HDM, and HDAC in conjunction with the conventional drugs for remission induction in ALL given at frequent intervals over 6 months. CR rates now range from 60%-100% compared to formerly 40% and LFS from 20%-65% compared to < 10%.27 Similar regimens are successfully administered in Burkitt lymphoma. It has become evident that an increase of methotrexate doses probably does not lead to further improvement of results but to a lower tolerability in adults. Some studies have included HDAC and achieved favorable results.28 Immunotherapy is also a promising approach in mature B-ALL since > 80% of the patients are CD20 positive (Table 5). Thus additional application of rituximab similarly as in high grade lymphoma may lead to an improvement of results.13,29
T-lineage ALL comprises the subtypes early T-ALL, thymic (cortical T-ALL), and mature T-ALL. It is characterized by a high WBC at diagnosis, mediastinal tumors (50%) and CNS involvement (8%) and a higher rate of CNS relapses (10%). T-ALL patients often have a large tumor mass and show rapid disease progression at diagnosis and at relapse. There are, however, few relapses after 3 years or more from diagnosis. In the GMALL studies the most relevant prognostic factor in T-ALL was the immunologic subtype with inferior LFS (< 30%) for early T-ALL and mature T-ALL compared to thymic (cortical) T-ALL (> 50-60%).30 T-ALL patients two decades ago had a poor prognosis in children as well as in adults. The median remission duration was 10 months or less, with an LFS < 10%. With recent treatment regimens CR rates of more than 80% and an LFS of 46% or more can be achieved in adults.13 C and AC are apparently important drugs in T-ALL.15 From childhood ALL studies there comes some evidence that HDM and A may be beneficial for consolidation therapy. Furthermore the inclusion of treatment elements with specific activity toward T-lymphatic blasts such as Cladribine, Campath, and arabinosyl-guanosine (Nelarabine) may contribute to further improvement of results.
Cytogenetics/Molecular Genetics
The most frequent cytogenetic aberrations in adult ALL t(9;22)/BCR-ABL (20-30%) and t(4;11)/ALL1-AF4 (6%) are associated with an inferior outcome. The prognostic impact of other aberrations is less clear. Thus 7, +8 and hypodiploid ALL were reported as unfavorable prognostic subgroups whereas a favorable outcome was detected for t(10;14) and a high hyperdiploid karyotype.31,32 The CALGB suggested a stratification into three prognostic subgroups: poor [including t(9;22), t(4;11), 7, and +8], normal diploid, and miscellaneous (all other structural aberrations), with LFS rates of 11%, 38%, and 52%, respectively.31 It remains, however, open whether cytogenetic abnormalities and also molecular aberrations such as p53, p15/16 mutations are independent prognostic factors since many of them are associated with certain immunologic subtypes.
Risk Stratification
The prognostic factors listed in Table 3 were applied in the current adult ALL trial of the GMALL study group. Patients without any risk factors were defined as standard risk (SR) and those with one or more risk factors as high risk (HR). Patients with Ph/BCR-ABL positive ALL were allocated to a separate very high risk group (VHR) since they are eligible for new and experimental treatment approaches such as tyrosine kinase inhibitors. According to this 48%, 33%, and 19% were allocated to the SR, HR, and VHR groups, respectively. The 5-year survival of CR patients was 55%, 36%, and 20% for the 3 groups. As a consequence, SR patients are not candidates for SCT in first CR in the GMALL studies whereas this is clearly indicated in patients with high-risk features. In other studies, however, all patients are treated uniformly, including allogeneic SCT in first CR for all patients with donor.2,4
Minimal Residual Disease
Methods, prognostic relevance and clinical application of MRD mainly in childhood ALL are extensively discussed in Section IV. In adult ALL a variety of potential clinical applications is already evident:13,33
Re-definition of CR: Molecular remission status in addition to remission status assessed by morphology.
Re-definition of new prognostic factors: MRD based risk factors in addition to conventional risk factors.
MRD-based treatment stratification: Individualized treatment with intensification in patients with high relapse risk and earlier stop of treatment in those with low relapse risk.
Molecular monitoring of new treatment elements: chemotherapy, antibody treatment, molecular therapy, SCT and others.
The evaluation of molecular CR rate of induction therapy and the risk stratification of MRD in the ongoing GMALL trial may serve as an example for practical application. A preliminary analysis of the ongoing GMALL study 06/99 demonstrated that the rate of molecular remissions as defined by negative MRD status with a sensitivity of < 104 is achieved in less than half of the patients with standard risk B-lineage ALL and in about 60% of the patients with T-lineage ALL. This correlates with the higher relapse risk in adult B-lineage compared to T-lineage ALL. In Ph/BCR-ABL positive ALL only 4% of the patients achieved a negative MRD status, which is in line with the very high relapse rate in this subgroup (Table 4 ).
Furthermore, MRD analysis at different time-points during the first year of chemotherapy demonstrated a strong correlation between the level of MRD and the relapse risk. It became evident that the combination of several time-points has the strongest prognostic impact. 52% of patients with MRD > 104 at one time-point from month 3 to 12 from diagnosis relapsed compared to no relapse in patients with MRD levels always below 104.34
According to these findings the GMALL group has started a prospective study (GMALL 06/99) with MRD based treatment decision after one year of chemotherapy.33,34 Patients with MRD < 104 at all time-points after induction are allocated to a MRD low risk (MRD-LR) group whereas patients with MRD > 104 at any time-point are defined as MRD high risk (MRD-HR). The remaining patients, e.g., patients without sensitive marker or inconclusive course of MRD are defined as intermediate risk (MRD-IMR).33 The major impact is that in MRD-LR patients treatment will be stopped after one year and maintenance therapy is omitted. Interim analyses of the study showed that MRD-HR patients can be identified already early during consolidation therapy and transferred to SCT.
New Treatment Options
Ph/BCR-ABL positive ALL
Ph/BCR-ABL positive ALL has an overall incidence in adult ALL of 20-25%, increasing with age to > 40% in patients above 50 years, and is the worst prognostic subgroup with an survival < 20%.36 Ph/BCR-ABL nearly exclusively occurs in B-precursor (c-/pre-B)-ALL. Diagnosis is made by cytogenetic analysis of the translocation t(9;22) and—with a higher sensitivity—by detection of the BCR-ABL rearrangement with PCR analysis. The CR rate for this group was improved to > 70% but is still somewhat lower than the 80-90% achieved for Ph/BCR-ABL negative common/pre-B ALL patients. However, < 30%37 up to only 4% (Table 4) of the CR patients are in molecular remission after intensive induction therapy depending on the sensitivity of the applied method. At present survival rates of 30-35% can be achieved with allogeneic SCT from sibling donors, although lower than for allogeneic SCT in CR1 in B-lineage Ph/BCR-ABL negative ALL. The tendency for the outcome of MUD transplants in small patient cohorts is even better (35-40%).38
Abl-tyrosine kinase inhibitor STI571
In Ph/BCR-ABL positive leukemia the bcr-abl fusion gene is causally involved in leukemogenesis and is considered to be essential for leukemic transformation. With a selective inhibitor of the Abl tyrosine kinase (STI571, Imatinib, Gleevec®) cellular proliferation of BCR-ABL positive CML and ALL cells can be inhibited selectively.39
In a first Phase II trial with STI571 in patients with relapsed/refractory Ph+ ALL monotherapy with STI571 induced a complete remission in 29% of the patients.40 The drug was particularly effective in patients with a relapse after an allogeneic SCT. The toxicity in these studies was comparable to the larger trials with CML patients. Gastrointestinal discomfort and nausea were the most frequent side effects, occurring in approximately 70% of patients, but nearly always mild (WHO grade 1 or 2). Clinical responses were correlated to BCR-ABL levels in bone marrow and peripheral blood.35 Thus quantitative PCR provides an option for continuous monitoring of the therapeutic effects of STI571. Based on these promising results Phase II studies in patients with de novo Ph/BCR-ABL positive ALL have been started. Several approaches such as treatment with STI571 in patients with MRD after induction therapy or after SCT or parallel to induction chemotherapy will be evaluated.
These studies have to evaluate the rate of cytogenetic and molecular CR and assess how durable it is. The effects in patients with MRD are of particular interest. In the future whether response rate and duration of response can be improved by combination therapies, e.g., with chemotherapy but also with other “molecular” drugs such as farnesyl transferase inhibitors, has to be evaluated. The detection of MRD will allow monitoring of treatment response and provides the option to evaluate effectivity of any combination treatment at short notice.
Antibody treatment
ALL blast cells express a variety of specific antigens such as CD20, CD19, CD22, CD33, and CD52 (Table 5 ) that may serve as targets for treatment with monoclonal antibodies (MoAbs). However, treatment with MoAbs is as yet not an established therapy in adult ALL. A prerequisite for Ab therapy is the presence of the target antigen on at least 20-30% of the blast cells. CD20, defined as expression on more than 20% of the blast cells, is expressed on more than one third of B-precursor ALL blasts, particularly in elderly patients (40-50%), and the majority of mature B-ALL blast cells (80-90%). This provides a rationale to explore the potential role of treatment with Rituximab (anti-CD20) in B-precursor ALL, mature B-ALL, and Burkitt’s lymphoma.
Other MoAbs [B43(Anti-CD19)-Genistein, B43(Anti-CD19)-PAP; Anti-B4-bR (Anti-CD19) in B-Lineage ALL and Anti-CD52 antibodies (Campath-1H), and Anti-CD7-Ricin in T-lineage ALL] have been investigated in Phase I-II pilot trials in ALL13 (Table 5). Also, antibodies developed for other diseases such as anti-CD22 in lymphoma and anti-CD33 in AML may be applicable in ALL since antigens are expressed in 17% and 16% of adult ALL cases, respectively. Antibody treatment could be administered as single agents or in combination with chemotherapy, for purging and as post-transplant therapy, and may be particularly effective in low-level disease (MRD-positive patients).
Microarray analysis
The use of microarrays for the identification of gene expression profiles is a new technology which already produced first clinically relevant results in adult ALL. Thus it was demonstrated that AML and ALL and also ALL subtypes (B- and T-lineage) can be distinguished by gene expression profiles. In childhood it was even possible to correlate molecular aberrations such as E2A-PBX1, TEL-AML1, MLL rearrangements, BCR-ABL, and high hyperdiploid ALL with distinct gene expression profiles.45
Microarray analysis was also used for identification of prognostic factors. In childhood ALL gene expression profiles differentiated between two prognostic subroups with LFS rates of 25% versus 98%.45 It was also possible to identify patients with resistance to STI571 exclusively according to their gene expression profile.46 These examples illustrate the exciting clinical applications resulting from this new method.
Beyond this another application of microarray technology, the analysis of differential gene expression, may serve to identify genes related to pathogenesis as recently demonstrated for T-lineage ALL. The expression of T-cell oncogenes (HOX11, TAL1, LYL1, LMO1, and LMO2) could be correlated to stages of T-cell development, which may in part explain the different outcomes of T-ALL subtypes.47 This method may contribute to the identification of new prognostic factors but also potential targets for molecular therapies.
II. Long-term Sequelae of Childhood Acute Lymphoblastic Leukemia
St. Jude Children’s Research Hospital, and Colleges of Medicine,
University of Tennessee Health Science Center, Memphis, TN
C-H. Pui is the American Cancer Society F.M. Kirby Clinical Research Professor.
This work was supported in part by grants CA 51001, CA 78224, and CA 21765 from the National Cancer Institute; the NIH/NIGMS Pharmacogenetics Research Network and Database (U01 GM61393, U01GM61374, http://pharmgkb.org/</index.html) of the National Institutes of Health; a Center of Excellence grant from the State of Tennessee; and the American Lebanese Syrian Associated Charities (ALSAC).
Contemporary intensive therapy cures at least 75% of children with ALL.1– 3 As the number of survivors of ALL increases, the late sequelae of treatment become increasingly important. The evolution of treatment strategies has also changed the spectrum of late complications. Although once prevalent, radiation-related complications (Table 6 ) are gradually being replaced by those associated with intensive chemotherapy. Long-term follow-up of survivors treated decades ago has also revealed some very late sequelae. This review focuses mainly on the complications that have recently emerged in survivors of childhood ALL.
Second Neoplasm
There are two main types of second neoplasms—acute leukemia (or myelodysplastic syndrome) and solid tumors. The latency period between treatment and the development of a secondary leukemia is generally short, whereas that of a secondary solid tumor is substantially longer. In fact, the risk of secondary solid tumor continues to rise for two or more decades after treatment.4
Incidence of second cancer
In a study conducted by the former Children’s Cancer Group, 8831 patients were treated for childhood ALL between 1983 and 1995. Of those, 63 developed second neoplasms, of which 39 were solid tumors, 16 were myeloid leukemia or myelodysplasia, and 8 were non-Hodgkin lymphoma.5 The cumulative incidence of any second neoplasm at 10 years from diagnosis of ALL was 1.18% (95% confidence interval [C.I.], 0.8-1.5%), which represents a 7.2-fold higher risk than that of the general population. The types of second neoplasm with increased risk included acute myeloid leukemia (AML), parotid gland tumor, thyroid cancer, brain tumor, soft tissue sarcoma, and non-Hodgkin lymphoma. In a study of 1597 patients treated on Dana-Farber Cancer Institute (DFCI) Consortium protocols between 1972 and 1995 (median follow-up, 7.6 years), the estimated cumulative risk of second neoplasm at 18 years was 2.7% (95% C.I., 0.7-4.7%).6 German investigators reported a 15-year cumulative risk of second neoplasm of 3.3% (95% C.I., 1.6-4.2%) in a cohort of 5006 patients treated in five consecutive Berlin-Frankfurt-Münster trials between 1979 and 1995 (median follow-up, 5.7 years).7 This risk was 14 times the risk of cancer in the general population. None of these studies included adequate follow-up of patients beyond 20 years.
In our recent review of patients enrolled in the first 12 St. Jude Total Therapy studies between 1962 and 1991, we observed a continual rise in the cumulative risk of second neoplasm beyond 20 years (unpublished data). In fact, among the 10-year event-free survivors, the cumulative risk of second neoplasm was 23.4% ± 4.0% (S.E.) from 10 years to 30 years after initial diagnosis of ALL. Virtually all of the second neoplasms were related to cranial or craniospinal irradiation, and most were benign or low-grade malignancies such as basal cell carcinoma, meningioma, or carcinoma of the thyroid or parotid gland. Among the patients who did not receive CNS irradiation, there was no increased risk of second neoplasm beyond 10 years after diagnosis of ALL. Our result underscores the importance of vigilance in monitoring long-term survivors of cancer, especially those who have received radiation therapy.
Therapy-related risk factors
Chemotherapeutic agents. Topoisomerase II inhibitors, especially etoposide and teniposide, and alkylating agents can induce the development of secondary hematopoietic neoplasms. More recently, therapy-related AML has been reported in patients, especially those with thiopurine methyltransferase (TPMT) deficiency, who were treated primarily with antimetabolite-based therapy.8
Topoisomerase II inhibitors stabilize the enzyme-DNA covalent intermediate and decrease the religation rate, thereby increasing DNA cleavage (damage) and subsequent cell death by apoptosis. If the DNA damage is insufficient to kill the cells and DNA recombination generates leukemogenic gene fusions, then malignant transformation can occur. Topoisomerase II inhibitor–related leukemias are characterized by balanced chromosomal translocations and short latency periods (2 to 4 years).9 Most cases of secondary leukemia related to the use of epipodophyllotoxins are characterized by balanced translocations involving the MLL gene at chromosome band 11q23 (at the breakpoint cluster region between exons 5 and 11) and one of the more than 40 partner genes identified to date.10 In contrast, cases of secondary leukemia related to anthracycline therapy often have other translocations, including 21q22 translocations, inv(16), t(15;17), or t(9;22).11 Topoisomerase II inhibitor-related secondary leukemias seldom have a preceding myelodysplastic phase.9 Although most cases of leukemia related to topoisomerase II therapy are AML, 10% are ALL.9
The risk of epipodophyllotoxin-associated leukemia is related to the schedule (i.e., the dose intensity) but not to the total dose.9 Two recent studies have demonstrated the emergence of a leukemia clone with MLL rearrangement 1.5 to 6 months after treatment with only three doses of topoisomerase II inhibitors.12,13 Concomitant treatment with alkylating agents, thiopurines, L-asparaginase, or radiation may potentiate the leukemogenic effects of topoisomerase II inhibitors.9 More recently, we found that short-term use of granulocyte colony-stimulating factor may also increase the risk of this complication (unpublished data). The outcome of treatment for topoisomerase II inhibitor–related secondary leukemia is dismal: only approximately 10% of these patients survive after chemotherapy, and 20% survive after hematopoietic stem cell transplantation.10 Although most oncologists recommend hematopoietic stem cell transplantation for this complication, the superiority of this treatment over chemotherapy has yet to be established, because none of the studies were adjusted for time to transplantation or for the patients’ characteristics (e.g., remission status, leukemic cell burden, and clinical condition).
By alkylating cellular molecules and DNA, alkylators cause inaccurate base pairing and single- and double-strand breaks in the double helix. Formation of interstrand crosslinks in DNA probably interferes with the orderly segregation of chromosomes at anaphase, thereby leading to the loss of genetic material. Thus, deletion of part or all of chromosomes 5 or 7 and their putative tumor-suppressor genes is a common finding in alkylator-related secondary leukemia. The incidence of this leukemia peaks 4 to 6 years after exposure and plateaus after 10 to 15 years.4 Leukemogenic potency differs among the alkylating agents, and cyclophosphamide is among the least potent.4 Higher cumulative doses and older age at the time of exposure are risk factors.4 Early stem cell transplantation and the use of busulfan at a targeted level of systemic exposure in the preparative regimen may improve outcome by reducing the rate of mortality not related to relapse.14
Irradiation. Irradiation can cause most types of cancer, which generally develop within or adjacent to the radiation field. These cancers have a prolonged latency, typically 15 to 30 years.4 In general, highly malignant tumors (e.g., glioblastoma multiforme, high-grade astrocytoma) have a shorter latency period (5 to 14 years), whereas that of the benign or low-grade malignant tumors (e.g., meningioma, basal cell carcinoma, thyroid carcinoma, parotid gland cancer) is longer (15 years or more).15 The risk of secondary cancer is increased by higher doses of radiation and by younger age at the time of treatment.4,15 A recent study suggested that growth hormone replacement therapy increases the risk of irradiation-related solid tumors (brain tumor and osteosarcoma) in survivors of childhood ALL.16 The outcome of treatment for irradiation-related second cancer depends on the type and resectability of the neoplasm.
Host-related risk factors
Microsatellite instability has been reported in cases of therapy-related leukemia or secondary tumor, a finding that suggests that an inherited defect in mismatch repair genes may play a role in the development of these complications.4 We and others8,17 have shown that patients with a deficiency of thiopurine S-methyltransferase, an enzyme that catalyzes the inactivation of thiopurines (6-mercaptopurine and 6-thioguanine), are at increased risk of therapy-related leukemia, especially if they have received concomitant epipodophyllotoxin treatment. These patients are also at increased risk of irradiation-related brain tumor when intensive antimetabolite therapy is given before and during cranial irradiation.18 Apparently, antimetabolites can potentiate the carcinogenic effects of epipodophyllotoxins and irradiation. In this regard, thioguanine in DNA can enhance topoisomerase II– mediated cleavage of DNA in the absence or presence of etoposide.19
Genetic polymorphisms of a number of drug-metabolizing enzymes are associated with the development of therapy-related leukemia or myelodysplasia. Glutathione S-transferases (GSTM1, GSTT1, and GSTP1) detoxify potentially mutagenic and toxic DNA-reactive electrophiles by conjugating them with glutathione. Inheritance of at least one valine allele at GSTP1 codon 105 was associated with an increased risk of chemotherapy-related leukemia.20 Presumably, a decrease in the enzyme activity increases the carcinogenic effects of the chemotherapeutic agents. NAD(P)H:quinone oxidoreductase (NQO1) is another detoxifying enzyme that reduces carcinogenic benzoquinones to less toxic hydroxyl metabolites. An NQO1 polymorphism that results in loss of the enzyme activity has been associated with increased risk of therapy-related leukemia and myelodysplasia.21,22 The cytochrome P450 (CYP) 3A4 metabolizes many chemotherapeutic agents, including epipodophyllotoxins, and generates DNA-damaging epipodophyllotoxin quinone metabolites. Patients with the CYP 3A4*1B variant allele have a decreased risk of topoisomerase II inhibitor–related leukemia; this reduced risk is putatively caused by the decreased production of reactive metabolites.23
By using DNA microarray analysis, we recently found that the presence of a certain gene expression profile of leukemic lymphoblasts at the time of diagnosis is predictive of therapy-related leukemia.24 Preliminary analysis showed that leukemic cells of patients with therapy-related leukemia overexpress the genes that encode MSH3, a mismatch repair enzyme, and RSU1, a suppressor of the RAS signaling pathway. Additional studies, especially those of normal host cells, may identify an expression profile that distinguishes individuals who are at risk of therapy-related cancer; these patients can then receive therapy modified to minimize carcinogenic agents. Alternatively, individuals at low risk or at no risk can be given highly effective but carcinogenic agents such as epipodophyllotoxins, thereby increasing the likelihood that their primary malignancy will be cured.
Endocrinopathy
Endocrine complications of therapy for ALL are common and potentially debilitating both during and after therapy. Acute complications such as adrenocortical insufficiency, diabetes mellitus, and the syndrome of inappropriate secretion of anti-diuretic hormone, and certain late effects, such as gonadal dysfunction and infertility, have been recently discussed elsewhere25,26 and will not be reviewed here.
Growth hormone deficiency
Growth failure is common in children treated for ALL and has been attributed to the interplay of multiple factors, including patient characteristics (e.g., age and sex), treatment variables (e.g., prior cranial irradiation), and post-treatment complications (e.g., graft-versus-host disease after hematopoietic stem cell transplantation). Growth hormone deficiency is observed primarily in patients who have been treated with a higher dose (>18 Gy) of cranial irradiation, and this deficiency appears to be more pronounced in girls who were very young (≤ 4 years of age) at the time of treatment.26 Early onset of puberty is another effect of cranial irradiation that limits the growth potential,25 especially in girls irradiated at a young age (i.e., < 4 years). A higher total dose of cranial irradiation (> 30 Gy) in patients who require a second course of irradiation for CNS relapse is generally associated with early onset of hormone deficiency within the first 5 years after treatment, whereas this complication may not develop for 10 or more years in those who receive a lower dose (18 to 24 Gy). In a study of adult survivors of childhood ALL who had received cranial irradiation (18 to 25 Gy), the deficiency of growth hormone became more pronounced with longer follow-up.27 Patients who received 18 Gy appeared to have a lower risk of developing growth hormone deficiency than did those who received 24 Gy or more.26 In a preliminary study, patients who received a reduced dose of cranial irradiation (12 Gy) and those treated with chemotherapy alone had the same growth velocity during the first 3 years after diagnosis.28
Growth hormone deficiency not only diminishes growth velocity and final height but also causes many complications, including dyslipidemia, decreased lean body mass, obesity, osteopenia, and reduced quality of life.25,26 Currently, growth hormone replacement therapy is given primarily to survivors during childhood to promote increased final height. However, the continuation of growth hormone replacement after the survivor attains final height should be considered, because the cessation of hormone therapy may have adverse consequences similar to those seen in patients with growth hormone deficiency (i.e., unfavorable body composition, osteopenia, adverse lipid profile, and reduced quality of life).25,29
The results of the recent Childhood Cancer Survivor Study suggested a possible association between growth hormone replacement therapy and the development of osteogenic sarcoma or meningioma within the irradiation field.16 Interestingly, both neoplasms express receptors for growth hormone and insulin-like growth factor-1, and their growth can be altered by manipulating these hormones.30,31 However, it should be noted that even if the increased risk of second cancer with growth hormone replacement therapy can be confirmed, the absolute number of second neoplasms is small (3 to 4 per 1000 person years at 15 years after diagnosis).16 Hence, the potential benefits of growth hormone replacement therapy probably outweigh the small risk of second neoplasm. With the trend toward reduction or elimination of cranial irradiation in the treatment of ALL,32–,34 this dilemma may one day be of historical interest only. Finally, growth hormone replacement therapy does not affect the risk of leukemic recurrence or survival.16,35
Obesity
Obesity is common in survivors of childhood ALL. Cranial irradiation (≥ 24 Gy), familial predisposition, and female sex are recognized risk factors for this complication.36–,38 Obesity is an important late effect, not only because of its associated morbidity and mortality, but also because of the associated social, psychological, and economic burdens.38 The mechanism responsible for increased adiposity after cranial irradiation is not known; however, the following mechanisms have been suggested: growth hormone deficiency, leptin insensitivity, and damage to the regions of the CNS that control appetite and eating behavior.25,39,40 Octreotide therapy dramatically reduced the rate of weight gain in two obese patients who had been treated with 18 Gy of cranial irradiation at ages 3 and 4 years;39 if confirmed, this finding may provide a therapeutic option for patients with hypothalamic obesity. The reduction or omission of cranial irradiation should reduce the prevalence of this complication in the current cohort of patients.
Bone demineralization
Osteoporosis has been observed in approximately 10% of pediatric patients at the time of diagnosis of ALL and in 67% or more of these patients during therapy.41 This complication may persist for 20 years or more after completion of therapy. Several risk factors have been associated with this complication, including corticosteroid and methotrexate therapy, cranial irradiation, decreased physical activity, and nutritional deficiency (altered metabolism of calcium, vitamin D, and magnesium).41 We recently identified male sex and white race as additional risk factors.42 Moreover, we found that cranial irradiation had a significant effect in patients who received 24 Gy but not in those who received 18 Gy.42 We hypothesized that a 24-Gy dose of cranial irradiation has a more profound effect on hypothalamic–pituitary function and that the resultant growth hormone deficiency and central hypothyroidism lead to diminished bone mineral accumulation. Strikingly, a recent DFCI study of 176 pediatric patients with ALL revealed that the 5-year cumulative incidence of fracture was 28% ± 3%.43 Male sex, age 9 years or older at diagnosis of ALL, and dexamethasone treatment were associated with a particularly high risk of fracture.
Most accumulation of bone mineral occurs between puberty and 25 years of age, after which little, if any, subsequent accretion occurs. Therefore, early intervention is necessary to prevent severe osteoporosis and its associated complications later in life. Patients should be encouraged to participate in weight-bearing activity. Vitamin D supplementation may not be appropriate during chemotherapy, because it does not prevent glucocorticoid-induced bone demineralization and could, in fact, increase the risk of hypercalcemia, urine calcium excretion, and urolithiasis, especially when glucocorticoid is included in the therapy.26 We are currently studying the effectiveness of oral calcium carbonate and vitamin D supplementation after completion of chemotherapy. Studies are also needed to determine the safety and effectiveness of treating children with calcitonin or bisphosphonates.
Avascular Necrosis of Bone
Avascular necrosis of bone or osteonecrosis is a well-recognized complication of treatment for childhood ALL and is generally attributed to the use of glucocorticoids. Depending on the type of therapy administered and the imaging methods used to detect this complication, the reported frequency of osteonecrosis varies from 0% to 40%.44 Virtually all of the joints can be affected, but the most common sites of involvement are the weight-bearing joints (hip, knee, and ankle).45 This complication is more common in girls who are more than 10 years of age, and it is more common in white patients than in African-American patients.
The increased risk of osteonecrosis in female patients may be related to their early pubertal development, because maturing bones (with epiphyseal closure and reduced intramedullary blood flow) are more susceptible to this complication. Factors contributing to the ethnic difference are unknown. We and others have recently decreased the duration of exposure to glucocorticoids during reinduction and continuation therapy, because the preliminary results of the Children’s Oncology Group suggest that intermittent rather than continuous use of dexamethasone reduces the risk of this complication.46
We are conducting a prospective study to determine whether debilitating complications of osteonecrosis can be prevented by early detection (monitoring with magnetic resonance imaging) and early intervention with decreased or omission of glucocorticoid therapy and physical therapy. Indeed, our preliminary results indicate that some early osteonecrotic changes are reversible with proper management.
Other Complications
Depending on the treatment regimens, some patients may develop cardiomyopathy or have impairment of neuropsychological performance. It is well recognized that high cumulative dose of anthracycline, female gender, young age and early onset of cardiotoxicity are risk factors for the development of late cardiomyopathy.47 The most commonly used screening tests for anthracycline-induced cardiomyopathy are echocardiography and radionucleotide ventriculography. A recent study suggested that plasma levels of natriuretic peptides may be useful markers for early detection of the complication.48 Because even a low cumulative dose of anthracycline can cause late cardiomyopathy,49 attempts have been made to reduce the toxicity by modifying the delivery schedule and by the use of cardioprotectant. However, in the Dana Farber 91-01 study, continuous infusion over 48 hours instead of bolus infusion did not appear to reduce the cardiotoxicity.50 Several ongoing studies are testing whether dexrazoxane can prevent short-term as well as long-term cardiotoxicity and whether this agent would reduce the antileukemic efficacy of anthracycline-containing treatment regimens.
By limiting the use of cranial irradiation and the judicious use of high-dose methotrexate, patients treated with contemporary therapy have far less neurotoxicity compared to those treated 10 to 15 years ago.51 However, even intrathecal therapy and dexamethasone can cause neuropsychologic deficits.52,53 Clearly, neuropsychologic function should be assessed in all survivors of childhood ALL, and appropriate remediation programs should be developed to address specific cognitive impairments.
III. Hematopoietic Cell Transplantation for Acute Lymphoblastic Leukemia
Frederick R. Appelbaum, MD*
Fred Hutchinson Cancer Research Center and University of Washington School of Medicine; FHCRC, 1100 Fairview Avenue North, D5-310, PO Box 19024, Seattle, WA 98109-1024
This work was supported, in part, by grants CA-18029 and CA-15704 from the National Cancer Institute, National Institutes of Health, DHHS.
With modern chemotherapeutic approaches, most children and a substantial proportion of adults with newly diagnosed ALL can be cured. Hematopoietic cell transplantation (HCT) plays an important role in the management of those patients who fail chemotherapy or are very likely to do so. This role continues to evolve as the outcomes of chemotherapy and transplantation change, sources of hematopoietic stem cells expand, and our ability to predict outcome improves.
Outcome According to Disease Stage
Primary induction failure
Patients who fail initial induction attempts are rarely, if ever, cured with subsequent chemotherapy. HCT has been reported to cure 10-20% of such patients.1,2 Because the window of opportunity to transplant patients failing induction is brief, strong consideration should be given to HLA typing all high-risk patients and their families at diagnosis.
After second relapse
Similar to the setting of primary induction failure, HCT offers the only prospect for cure for patients with ALL whose disease has advanced beyond second remission. Long-term disease-free survival (DFS) has been reported in 10-25% of such cases with HCT, with posttransplant relapse being the dominant reason for failure.3– 6
Second remission
For children, HCT is appropriate therapy for those who relapse while on therapy or within 6 months of completing it. While truly randomized trials have not been performed, careful case-controlled analyses have. An International Bone Marrow Transplant Registry(IBMTR)/Pediatric Oncology Group study comparing 255 matched pairs reported a 40% DFS with transplant compared to 17% with chemotherapy.7 A German cooperative study reported 56% DFS with HCT compared to 22% with chemotherapy.8 A Nordic case-controlled study similarly found an advantage in event-free survival at 3 years for transplantation (40%) versus continued chemotherapy (23%).9 Similarly, the Italian Bone Marrow Group and the Italian Pediatric Hematology/Oncology Association reported significantly longer DFS with transplantation compared to chemotherapy for children with ALL in second CR.10 Smaller, single institution studies have reached similar conclusions.11
Few adults with ALL who suffer an initial relapse can be cured with chemotherapy. Recent results from the IBMTR report on 388 adults age > 20 with ALL in second remission transplanted from HLA-identical siblings between 1994-1999. The probability of survival at 5 years is 30%.12 Results using matched unrelated donors in 215 adults are very similar. These outcomes are almost certainly better than chemotherapy can achieve and, thus, transplantation can be considered for most adults with ALL in second remission.
First relapse
Occasionally, patients with acute leukemia are identified in early first relapse, raising the question of whether one should proceed directly to transplant or whether reinduction should first be attempted. In the setting of AML, there are data suggesting that a strategy of immediate transplantation for patients in early relapse is reasonable if feasible. In contrast, there are virtually no data describing the outcome of allogeneic transplantation for ALL in untreated first relapse.
First remission
Because modern chemotherapeutic regimens cure 70-80% of children with ALL, there is no role for HCT in patients with standard risk disease. There are, however, some children with very high risk disease for whom transplantation in first remission is appropriate. Children with Ph+ ALL appear to benefit from transplantation in first CR. A retrospective review of 326 children with Ph+ ALL treated by 10 study groups or large institutions from 1986 to 1996 found an advantage for transplantation in first remission compared to chemotherapy for children in all Ph+ risk groups.13 Similar findings, albeit in smaller numbers of Ph+ ALL children, have been reported from the Tokyo Children’s Cancer Study Group and others.14,15 In contrast, although children with t(4;11) also do poorly with chemotherapy, there is less evidence that transplantation benefits these children.16 When a broader definition of high-risk ALL is used, results have been more difficult to interpret, with some studies finding a strong benefit of transplantation17,18 and others finding no benefit.19 Given the biologic heterogeneity of high-risk ALL, it is not surprising that results might vary from study to study when too broad a definition of high risk is used.
The indications for allogeneic transplantation for ALL in adults in first remission are also evolving. Results with standard chemotherapy regimens generally report cure rates in the 35-45% range, while data from the IBMTR report approximately 50% survival for transplantation in 909 adults with ALL transplanted in first CR. Earlier attempts at case-controlled studies performed by the IBMTR showed no overall advantage for transplant over chemotherapy.20,21 However, reexamination of this issue using more recently treated patients demonstrates superior DFS with transplantation for patients less than age 30.22 The sole published prospective trial comes from the French LALA group and involved patients ages 15 to 40 who, once they had achieved a CR, were assigned to allogeneic transplantation if they had a matched sibling or randomized to autologous transplantation versus chemotherapy if they did not. When patients were analyzed according to protocol intent, an advantage was found for allografting versus chemotherapy or autografting patients (DFS 46% versus 31%).23 High-risk patients, defined as Ph+, age > 35, white blood cell count > 30,000/mm3 at diagnosis, or time to achieve remission > 4 weeks, particularly benefited from allografting (DFS 44% versus 11%). Immunophenotyping, and in particular, coexpression of lymphoid and myeloid antigens on the same cell (biphenotypic) or in two different populations (hybrid leukemia) was not included in this model and has not been a consistent predictor of high-risk disease.
Central Nervous System Disease
Presence of CNS involvement at diagnosis does not appear to be an adverse risk factor for either children or adults. Children with an isolated CNS relapse can still do very well if treated with systemic reinduction chemotherapy together with re-treatment of the CNS. In contrast, adults with isolated CNS relapse do much more poorly. If such patients are referred for transplantation, it is important that their CNS disease be brought into control before the transplant since the preparative regimen alone is seldom capable of eradicating CNS disease.24,25 CNS therapy with methotrexate plus cytarabine, or depo-cytarabine should be considered.
Hematopoietic Stem Cell Source
Genetic relationship
The hematopoietic stem cell source can be defined according to the genetic relationship between the donor and recipient and according to the anatomic source of the stem cells. A central question surrounding the choice of donor is whether there is any evidence for a graft-versus-leukemia (GVL) effect in ALL. Although studies performed more than two decades ago suggested such an effect, more recent reports demonstrating a relative lack of efficacy of donor lymphocyte infusions (DLI) in ALL have called the existence of a GVL effect in ALL into question. A recent study involving 1132 patients with T- or B-lineage ALL confirmed earlier observations that both acute and chronic graft-versus-host disease (GVHD) are associated with lower overall risks of relapse and that this effect is similar in T- and B-lineage ALL.28
In general, HLA matched siblings are the donors of choice. Use of family members who are mismatched with the patient at a single class I or class II antigen generally results in survival similar to that seen with matched siblings albeit with a somewhat higher incidence of GVHD or graft rejection. Use of family donors mismatched at more than one HLA antigen is generally associated with a substantial increase in both GHVD and graft rejection so that overall survival has been distinctly worse. Attempts to improve outcome using two or three antigen mismatched family members have included T cell depletion coupled with infusion of large doses of CD34 selected hematopoietic cells and/or enhanced myeloablative preparative regimens to help prevent graft rejection. While some encouraging results have been reported, the number of patients with ALL involved in any one trial is still quite limited and results are very preliminary.29,30
The use of matched unrelated donors offers an alternative to the use of mismatched family members. While unrelated donor grafts are generally associated with more GVHD and other complications than matched sibling transplants, the compensatory decrease in relapse rates plus recent improvements in supportive care have narrowed the gap between the two approaches. Recent single institution or group studies dealing with both children and adults have reported outcomes using unrelated donors that are not much different from that seen with matched sibling transplants. For example, the Nordic Transplant group has reported similar outcomes using matched siblings and unrelated donors in both children and adults.31,32 Overall results reported by the IBMTR involving 4241 transplants for ALL performed between 1991-1997 report a DFS of 44% for patients using matched unrelated donor transplants in first remission versus 52% for patients in first remission undergoing transplantation from matched sibling donors, and of 35% versus 42% for patients in second remission, again using matched unrelated donors versus matched sibling donors.12 The degree to which patient selection contributes to the above noted differences is unknown. The extent to which mismatching among unrelated donor recipient pairs effects outcome is now becoming clearer. While mismatching at a single class I allele can increase the risk of graft rejection and mismatching at a single class II allele seems to increase the risk of GVHD, the overall impact of single antigen mismatching on overall survival does not appear to be significant.33,34 However, if more than one mismatch exists between donor and recipient, overall results appear to deteriorate, with diminished overall survival.
The role of autografting in ALL remains largely undefined. The IBMTR reports 43% survival at 3 years after autografting for ALL in first remission. However, in the only randomized trial reported to date, there was no advantage to autografting compared to continued chemotherapy for patients in first CR.23 The IBMTR also reports a 37% survival at 3 years for patients autografted in second remission, a surprisingly good result. One case-controlled study has compared 214 patients receiving autografts for ALL compared with 337 similar patients receiving unrelated donor transplants. Among those transplanted in second remission, unrelated donor recipients had a superior disease-free survival.26
Anatomic source
While bone marrow has traditionally been the source of stem cell for matched sibling allogeneic transplantation, pilot and Phase II studies published in the mid-1990s suggested that use of G-CSF mobilized peripheral blood cells led to rapid engraftment without an apparent increase in acute GVHD.35,36 A recent prospective randomized trial demonstrated not only that use of peripheral blood led to faster engraftment without an increase in acute GVHD but that overall survival was improved with the use of peripheral blood versus marrow. Although the study was not prospectively sized to look at relative effects of peripheral blood in subpopulations of patients, the advantage of peripheral blood was more apparent in patients at high risk of relapse compared to low-risk groups.37 Retrospective analyses have generally been consistent with the results of this prospective randomized trial.38 There is less data concerning the use of mobilized peripheral blood for unrelated donor transplants. Phase II data seem consistent with findings in the matched sibling setting, that is, faster engraftment without a clear increase in acute GVHD, but randomized trials have not been reported.39
Previously cryopreserved unrelated cord blood offers an alternative to unrelated donor transplants. Potential advantages include relatively rapid availability and, because cord blood is relatively deficient in T cells, the possibility that increased degrees of mismatching might be tolerable. However, cord blood has also been associated with slower engraftment and an increased incidence of graft failure and late fatal infections. In the largest study published to date, Rubinstein et al reported the outcome after cord blood transplant and found it significantly influenced by the number of cells per kilogram infused as well as the patients’ underlying disease, age, and degree of match with the donor.40 Two retrospective studies have attempted to compare outcomes using unrelated cord blood versus unrelated marrow for transplantation of children with acute leukemia. While a single institution study involving 114 children found no significant difference in survival, a larger multi-center study involving 541 patients found a 2-year DFS of 43% for unrelated marrow versus 31% for cord blood.41,42
Preparative Regimens
The most commonly used preparative regimens prior to allogeneic transplantation for ALL consist of cyclophosphamide (CY) plus total body irradiation (TBI) with or without the addition of either etoposide or cytarabine. As in most other transplant settings, there have been few studies comparing different preparative regimens. A retrospective analysis from the IBMTR found that the conventional CY/TBI regimen was superior to a non-TBI containing regimen of busulfan (BU) plus CY, with 3-year survival of 55% with CY/TBI versus 40% with BU/CY.43 Curiously, the risk of relapse was similar in the two groups while non-relapse mortality was higher in the BU/CY group. Only a limited number of new concepts are being tested to improve the anti-tumor efficacy of preparative regimens. Our group in Seattle has been studying the use of a radiolabeled anti-CD45 monoclonal antibody as a method of delivering additional radiation specifically to the marrow, spleen, and lymph nodes as part of a transplant preparative regimen.44
Interest in the use of less aggressive preparative regimens in the treatment of ALL has been fueled by continued awareness of a GVL effect with transplantation and increased appreciation that engraftment can be achieved with less than fully myeloablative preparative regimens. Initial studies of so-called nonmyeloablative transplants have generally been conducted in patients who are not candidates for conventional transplants because of age or comorbid conditions. A variety of different regimens have been used ranging from the minimum necessary to achieve engraftment (fludarabine plus very low-dose TBI)45 to regimens of more intermediate intensity (fludarabine plus melphalan, for example).46 Reports to date show the feasibility of the approach with the large majority of patients engrafting and, as expected, fewer immediate toxicities than seen with ablative regimens. Complete responses have been documented in patients with a variety of hematological malignancies. However, because these transplants have been performed for older patients, there is as yet very little data about the use of this approach for patients with ALL.
Monitoring Minimal Residual Disease
Molecular techniques that allow for the detection and quantitation of small amounts of tumor cells in patients who are in clinical remission have been used to study patients both posttransplant, and more recently, pretransplant. Radich et al showed that patients with Ph+ ALL who are polymerase chain reaction (PCR) positive posttransplant have a far higher likelihood of relapsing than patients who are PCR negative.47 Of particular interest was the observation that patients PCR positive for p190 posttransplant had an 88% chance of relapse compared to a relapse rate of only 12% for patients PCR positive for p210 posttransplant. A more recent update of this question in 90 Ph+ ALL patients transplanted in Seattle showed a 5-year survival of 29% in patients who were PCR positive at any time posttransplant compared to 57% in patients who remained PCR negative (Stirewalt, submitted). Similar to the story with Ph positivity, detection of the clonal V-D-J immunoglobulin gene rearrangement by PCR during the first 100 days posttransplant is associated with a higher incidence of relapse compared to PCR negative patients.48 While no study has yet shown that early interventions based on the detection of these sorts of markers is of clinical benefit, such approaches are under study.
An additional use of monitoring minimal residual disease may be to assess the likelihood of a successful transplant based on the degree of disease immediately pretransplant. Knechtli et al reported on 64 children with ALL in first or subsequent remission undergoing allogeneic transplantation. Minimal residual disease was measured as high, low, or non-detectable during the month prior to transplant, and the 2-year event-free survival rates posttransplant for these three groups were 0%, 36%, and 73%, respectively.49
Treatment of Posttransplant Relapse
Patients who relapse following allogeneic transplantation for ALL have a very poor prognosis. While efforts to manipulate the immune system by withdrawing immunosuppression or adding DLI have met with some success in patients with chronic myeloid leukemia, chronic lymphocytic leukemia, and other selected malignancies, the response rates to DLI in patients with active ALL have been very low, in general less than 20%.50,51 It is uncertain whether these low response rates reflect primarily the pace of ALL growth or that ALL cells are poor targets for immunotherapy. An obvious approach to attempt to improve the outcome of DLI is to infuse cells only after reinduction with chemotherapy. There is some suggestion that administration of IL-2 may induce responses in patients who have failed to respond to initial DLI.52
Patients who relapse following an autologous transplant have occasionally been treated with allogeneic transplants. In a small study from Seattle, the DFS 2 years from second transplant was 23%.53 Results were better in younger patients who had been treated back to remission before the second transplant.
Improving the Outcome of Transplantation for ALL
Any approach that improves the outcome of transplantation generally, such as better control of GVHD or infections, would obviously benefit transplants for ALL, but such generic approaches will not be discussed here. The development of improved preparative regimens for treatment of ALL has been considered earlier in this review. Other approaches specific to ALL include better timing of transplantation and development of approaches to specifically target ALL cells using a GVL reaction.
Overall outcome of treatment for ALL would almost certainly be improved if transplantation could be applied during first remission for every patient who is destined to fail initial chemotherapy treatment and be avoided altogether in those patients who have or will be cured by chemotherapy. As noted earlier, features identifying high risk patients include, age, white count at diagnosis, immunophenotype, cytogenetics, and rapidity of induction response. More recently, measurement of disease burden immediately after induction using multi-dimensional flow cytometry and continued monitoring of disease burden in patients in apparent complete remission using PCR approaches may allow for more accurate identification of patients destined to fail chemotherapy before overt relapse. The test then will be to see if early transplantation is of benefit to these very high risk patients. Measurements of minimal residual disease might also be used to alter the transplant approach. As noted earlier, use of PCR approaches to measure disease burden prior to transplant may allow for the identification of patients likely to relapse posttransplant. Uzunel et al have further shown that the development of acute and chronic GHVD may be of particular importance in preventing relapse in patients with high levels of MRD.54 Thus, one may want to, for example, give reduced doses of cyclosporine to such patients since it has been suggested by Locatelli et al that lower cyclosporine dosing may reduce the risk of relapse.55
While lowering cyclosporine dosing in patients at high risk for relapse or infusing donor lymphocytes for patients with persistent disease posttransplant may have some limited benefit, in general, efforts to improve overall outcome of allogeneic transplantation for ALL by such non-specific manipulations of the immune system have been disappointing (reviewed in ref. 56). Several approaches have been taken to try to specifically augment the GVL effect seen posttransplant without increasing GVHD. One general strategy has been to identify polymorphic minor histocompatibility antigens that are differentially expressed by hematopoietic and nonhematopoietic tissues. Such antigens should be able to serve as targets for donor-derived T cells administered posttransplant with the goal of ablating all normal and malignant hematopoietic cells of the host. A number of such antigens have been identified and clinical trials using this approach are already underway in Seattle and elsewhere.57,58 An alternative approach is to identify antigens associated with the malignant phenotype. Such antigens might be mutational (such as bcr/abl), viral (such as EBNA), or could be an overexpressed self antigen such as WTI, proteinase 3, or AF1q.59,60 A final approach has been to develop whole cell vaccines using approaches that augment the host tumor response. For example, pre B-ALL cells are thought to be poor immunogens because they lack B7-1. If ALL cells are transduced to express B7, they express self antigens much more effectively and with their use, it has been reported that it is possible to generate autologous T cell lines with relative specificity to the leukemia.61
IV. Minimal Residual Disease in Acute Lymphoblastic Leukemia: Improved Techniques and Predictive Value
Jacques J.M. van Dongen, MD PhD,*
Department of Immunology, Erasmus MC, University Medical Center Rotterdam, Dr. Molewaterplein 50, 3015 GE Rotterdam, The Netherlands
Dr. van Dongen has received support from BIOMED-2 and IVS Technologies.
Acknowledgments. The authors thank Prof. M. Schrappe, Prof. C.R. Bartram, Prof. G. Masera, Prof. A. Biondi, Prof. H. Gadner, and Prof E.R. Panzer-Grümayer of the International BFM Study Group, the members of the European Study Group on MRD detection in ALL, and the Board of the Dutch Child-hood Leukemia Study Group for pleasant collaboration as well as for fruitful and critical discussions during the last ten years.
The classical definition of remission in ALL, based on cytomorphology of bone marrow (BM), still permits the presence of up to 5% of lymphoblasts. Consequently, this provides only superficial information about the effectiveness of the treatment (Figure 1 ), because only a small proportion of patients (< 5% of children and ∼15% of adults with ALL) with a very poor prognosis fail to achieve cytomorphological remission. In contrast, within the patient group that achieves remission, morphology is unable to discriminate between patients at high risk of relapse and patients with excellent prognosis. Therefore, more sensitive techniques were developed during the last 15 years for detection of lower frequencies of malignant cells during and after treatment, i.e. detection of MRD. Several studies have proven that MRD monitoring in ALL patients has significant prognostic value, which can be used for improved therapy stratification.
At present such clinically relevant MRD information can be obtained in ALL with three different techniques (reviewed in ref. 1– 3):
flow cytometric immunophenotyping using aberrant or “leukemia-associated” phenotypes;
PCR techniques using chromosome aberrations that result in fusion gene transcripts or aberrant expression of transcripts;
PCR techniques using patient-specific junctional regions of rearranged immunoglobulin (Ig) and T-cell receptor (TCR) genes.
These three techniques are suitable for MRD detection because they are characterized by most of the following features:4
sensitivity of at least 10–3 (one malignant cell within 1000 normal cells), but frequently of 10–4 to 10–6;
applicability in the vast majority of patients under study;
leukemia-specificity (ability to discriminate between malignant and normal lymphoblasts, without false positive results);
intralaboratory and interlaboratory reproducibility;
feasibility (easy standardization and rapid collection of results for clinical application);
quantification (possibility of precise quantification of MRD levels).
MRD Techniques
The characteristics of the three currently used MRD techniques are summarized in Table 7 . Each of the three MRD techniques has specific advantages and disadvantages, which should be weighed against each other when plans are made for large-scale clinical MRD studies. Particularly, the required sensitivity and the applicability will play an important role, because these two characteristics determine which patients can actually be monitored.
Flow cytometric MRD detection
The immunophenotypic “targets” in flow cytometric MRD detection mainly concern aberrant or “leukemia-associated” immunophenotypes, which are rare (or sometimes absent) in normal BM and peripheral blood (PB) (reviewed in ref. 2,4). Current triple and quadruple labelings allow detection of such leukemia-associated phenotypes in the majority (60-98%) of precursor-B-ALL and in virtually all T-ALL.5–,8 Immunophenotypic shifts during the disease course do occur, although their reported frequency is variable, mainly because different definitions are used for phenotypic shifts.2,9 Nevertheless, most groups agree that preferably two different leukemia-associated phenotypes should be monitored per patient to prevent false-negative results.2,4
The sensitivity of flow cytometric MRD detection remains unclear. Flow cytometric evaluation of 106 cells is technically not a problem, but detection of 50 to 100 malignant precursor-B-cells between 10,00050,000 normal precursor-B-cells in BM is not easy, particularly in regenerating BM during or after therapy.10 However, this is essential to reach sensitivities of 104. Some experienced centers claim that a detection limit of 104 can be reached routinely in virtually all ALL patients, but other centers agree that the detection limit varies between 103 and 104 for most precursor-B-ALL, while in virtually all T-ALL a detection limit of 104 can indeed be reached, because of their specific thymocytic phenotype (Figure 1).
PCR analysis of chromosome aberrations
Structural chromosome aberrations are ideal leukemia-specific PCR targets, which remain stable during the disease course and can reach excellent sensitivities of 104 to 106. In ALL, these PCR targets mainly concern fusion gene transcripts (e.g., TEL-AML1, BCR-ABL, and SIL-TAL1; see Table 8 ) or aberrantly expressed specific transcripts (e.g., HOX11L2 and WT1), which can be detected via reverse transcriptase (RT) PCR analysis.3,11– 13
Two main disadvantages limit the application of chromosome aberrations as MRD-PCR targets: applicability in only a minority of ALL patients and the chance of false-positive results via cross-contamination of PCR products.
PCR analysis of Ig and TCR gene rearrangements
The junctional regions of rearranged Ig and TCR genes are fingerprint-like sequences, which differ in length and composition per lymphocyte or lymphocyte clone and consequently also per each lymphoid malignancy, such as ALL.14 These patient-specific MRD-PCR targets can be detected in the vast majority of precursor-B-ALL and T-ALL (Table 9 ) and generally reach sensitivities of 104 to 105.1,15– 17
These high sensitivities require the precise identification of the junctional region sequences of Ig and TCR genes in each ALL, because these sequences are needed to design patient-specific oligonucleotides. Subsequently, sensitivity testing has to be performed via serial dilution of DNA obtained at diagnosis in order to assess whether the required sensitivity can indeed be reached.
If only sensitivities of 102 to 103 are required, it is possible to skip the sequencing of the junctional regions and focus on differences in length of the junctional regions, which can be evaluated by PCR product length assessment via fluorescent GeneScanning. An internal competitor containing the same type of rearrangement can be used for quantification of the Ig/TCR gene target.18
The main disadvantage of using Ig/TCR gene rearrangements as MRD-PCR targets in ALL is the occurrence of continuing rearrangements during the disease course as has been identified by comparing the Ig/TCR gene rearrangement patterns at diagnosis and relapse.19,20 Such changes in rearrangement patterns will lead to false-negative PCR results.
In precursor-B-ALL, changes at relapse were particularly observed in patients with more than one leukemic subclone (oligoclonality) at diagnosis.20 The occurrence of subclones differs per type of Ig/TCR gene rearrangement and consequently also the stability differs per type of rearrangement (Table 9). For example, IGK gene rearrangements involving the so-called kappa deleting element (Kde) are more stable than IGH, TCRG, and TCRD gene rearrangements.20 The high stability of IGK-Kde rearrangements is most probably caused by the fact that these rearrangements not only delete the IGK constant region (Cκ), but also the two enhancers (iEκ and 3IEκ). This implies that Kde rearrangements are “end-stage” rearrangements, which cannot easily be subject of continuing rearrangements (Figure 2 ).15
In T-ALL, the changes in TCR gene rearrangement patterns at relapse are more limited, probably related to the fact that T-ALL rarely contains oligoclonal TCR gene rearrangement patterns.16,19 Nevertheless, it is now generally accepted that preferably two Ig/TCR gene targets should be used for reliable and sensitive MRD detection in ALL patients.
Real-time quantitative PCR techniques
PCR-based MRD methodologies are increasingly achievable thanks to the development of real-time quantitative (RQ-) PCR techniques (reviewed in ref. 21). In classic PCR analyses with end-point quantification, minor variations in primer annealing and extension may lead to major variations after 30-35 PCR cycles. In contrast, RQ-PCR permits accurate quantification during the exponential phase of PCR amplification. In RQ-PCR assays an amplification plot is generated, and the cycle at which the fluorescence signal exceeds a certain background fluorescence level (threshold cycle) is directly proportional to the amount of target RNA or DNA present in the sample. For MRD detection, a dilution series of the initial diagnostic sample can be made and the amount of residual leukemic cells in follow-up samples during or after treatment can be calculated by use of the standard curve of the initial diagnostic sample.
Fusion gene transcripts from chromosome aberrations or aberrantly expressed transcripts are excellent RQ-PCR targets for the detection of MRD in ALL.22,23 Copy numbers of the relevant transcript in BM or PB follow-up samples can be calculated via a dilution curve of known amounts of plasmids containing the fusion gene sequences. A standardized approach has been developed for the most frequent fusion gene transcripts in ALL via the Europe Against Cancer program.24
Several studies have proven that RQ-PCR can be effectively used for quantitative detection of MRD using junctional regions of Ig and TCR gene rearrangements as PCR targets. Uniform approaches with a junctional region-specific primer in combination with a germline TaqMan probe and primer have been developed for the classical Ig/TCR targets: IGH, IGK-Kde, TCRG and TCRD.25–,28 Introduction of new MRD-PCR targets like TCRB gene rearrangements is currently a subject of an European BIOMED-2 Concerted Action.29
Prognostic Value of MRD Detection in ALL
Early retrospective and small prospective studies with relatively short follow-up indicated that the detection of MRD in childhood ALL predicts treatment outcome, although the results of these clinical studies were not fully concordant (reviewed in ref. 1). This was attributed to differences in sensitivities of MRD monitoring as well as to differences in intensity of cytotoxic treatment protocols. Several recent large-scale prospective studies confirmed the clinical value of MRD monitoring, justifying incorporation of MRD information in current childhood ALL treatment protocols to refine risk assignment.5,8,30– 33
Significance of MRD detection during front-line treatment of childhood ALL
The most significant application of MRD monitoring for front-line treatment of ALL is the evaluation of the initial response to cytotoxic therapy. Low levels or absence of MRD in BM after completion of induction therapy appears to predict good outcome.5,8,30–,34 Depending on the treatment protocol and the end-of-induction sampling time point, MRD-negativity is associated with overall relapse rates of only 2-10% (Figure 3 and Table 10 ).5,8,30–,33 Moreover, sensitive MRD detection during the induction phase seems capable of identifying 20% of childhood ALL patients with a very rapid leukemia clearance and long-term relapse-free survival.35 On the other hand, several studies proved that high MRD levels at the end of induction treatment are associated with high relapse rates of 70–100% (Figure 3 and Table 10).5,8,30,32,36 Statistical analyses have shown that the MRD status after induction therapy is the most significant prognostic factor, independent of other clinically relevant risk factors, such as age, blast count, immunophenotype, presence of chromosome aberrations at diagnosis, and response to prednisone.30,32,37
Clinical value of MRD during treatment of relapsed ALL
The predictive value of MRD monitoring is particularly clear after ALL relapse, as shown by the BFM group.38 This can be perceived as assessment of early treatment response after second induction treatment. Low MRD levels (< 10-3) were associated with a probability of relapse-free survival of 86%, whereas MRD levels ≥103 were uniformly predictive of dismal outcome (probability of relapse-free survival of 0%; see Figure 4 ).38 Apparently, MRD information also has a high predictive value in relapsed ALL. However, the patient numbers are still small and the results need further confirmation.38
Clinical value of MRD detection before bone marrow transplantation in childhood ALL
MRD monitoring was shown to be very informative for ALL patients undergoing bone marrow transplantation (BMT).39–,43 In patients receiving T-cell depleted grafts, high levels of MRD-PCR positivity (102 to 103) before allogeneic BMT were invariably associated with relapse after transplantation.39,42,43 The 2-year event-free survival in patients with low level of MRD positivity (103 to 105) was 35 to 50%, irrespective of graft manipulation.39,42,43 It has been suggested that significant GVHD associated with non-depleted grafts might overcome MRD positivity, even high MRD levels.42,43 In contrast, MRD-negativity before allogeneic transplantation significantly correlated with better outcome and 2-year event-free survival higher than 70% (Figure 4).39–,43 Therefore, patients with a high MRD burden prior to BMT might be offered alternative treatment (e.g., further cytoreduction before BMT, intensified conditioning, and/or early post-BMT immunotherapy) in order to improve their generally poor outcome.39,41
Clinical value of MRD detection in adult ALL
The preliminary results of MRD studies in adult ALL showed molecular response to chemotherapy similar to childhood ALL, but with higher frequencies of persistent MRD positivity in adults.44,45 Not only the frequencies of MRD positivity, but also the MRD levels in adult patients were significantly higher than in comparably treated children, which is consistent with a higher in vivo drug-resistance of adult ALL.44,45 A single prospective MRD study in t(9;22) negative adult precursor-B-ALL patients demonstrated the strong predictive value of MRD information at all investigated time points within two years of treatment, particularly 3-5 months after remission induction and beyond.46
Recognition of MRD-based risk groups
The results of the large prospective MRD studies in childhood ALL indicate that MRD analysis at a single time point gives highly significant prognostic information, but a single time point is not sufficiently precise for defining MRD-based low-risk and high-risk groups.5,8,30–,32 Depending on the MRD study, the end-of-induction MRD status either identifies only patients at low risk of relapse30,31 or more frequently identifies exclusively high-risk patients (Table 10).32,37 In contrast, information on the kinetics of tumor load decrease, e.g., at the end of induction treatment and before consolidation treatment, appeared to be highly reliable for the recognition of all clinically relevant risk groups.30,33 As shown by the International BFM Study Group (I-BFM-SG), this combined MRD information distinguished patients at low risk with MRD negativity at both time points (5-year relapse rate of 2%); patients at high risk with intermediate (103) or high (≥102) MRD levels at both time points (5-year relapse rate of 84%); and the remaining patients at intermediate risk (5-year relapse rate of 24%) (Figure 3).30 The group of MRD-based high-risk patients was larger than any previously identified high-risk group (approximately 15%) and had an unprecedentedly high 5-year relapse rate of 84%. On the other hand, the MRD-based low-risk patients made up a group of a substantial size (approximately 45%), comparable to the frequency of survivors of childhood ALL in the 1970s before treatment intensification was introduced.47 Such a precise MRD-based risk stratification has not yet been demonstrated for adult ALL.
Can we compare the MRD results of different clinical ALL trials?
The prognostic value of MRD detection during the early treatment phases of childhood and adult ALL has been established. In fact, every published study has confirmed the high prognostic value of MRD data obtained at the end of induction treatment.5,8,30– 32 Nevertheless, the reported large-scale MRD studies show remarkable differences in the meaning of MRD level information at the end of induction (Table 10). Also the MRD-based risk groups are defined differently, resulting in different distributions of the patients over the risk groups and different relapse rates (Table 10).
The major differences in risk group definition and corresponding relapse rates might be related to the type of treatment protocol, the timing of the follow-up samples, or the applied MRD technique (e.g., sensitivity and/or stability of the applied targets). Consequently, it will be impossible to extrapolate data from one clinical treatment protocol to another. This implies that for each treatment protocol the “MRD window” has to be defined precisely: sampling time points versus required sensitivity. In practice, this means that MRD-based treatment intervention should always be designed according to earlier-obtained MRD results from the same treatment protocol.
How to translate MRD information into new clinical treatment protocols?
When MRD information from existing treatment protocols is translated into new clinical treatment protocols, several MRD-related aspects will influence the implementation of the new protocol:
MRD-based stratification can only be introduced in the protocol after the actual MRD measurements, i.e., 6 to 13 weeks after start of treatment.
The treatment blocks before the MRD sampling time points cannot be changed, because this would directly change the prognostic value of the MRD results.
Preferably at least two early MRD sampling time points should be used, because this results in a more accurate definition of MRD-based risk groups.
MRD-based high-risk and low-risk patients
Protocol Committees will choose for specific MRD-based aims in new protocols, depending on the results of the preceding clinical MRD studies (Table 10). MRD-based high-risk patients (with relapse rates of 75 to 100%) might profit from further treatment intensification including SCT or new treatment modalities. For example, this was a logical choice for the ongoing MRD-based BFM-AIEOP and EORTC studies (Figure 3).
MRD-based low-risk patients (with relapse rates 0-2%) represent a group of substantial size in childhood ALL and might profit from treatment de-intensification, e.g., reduction of re-intensification treatment, as aimed for by the current BFM-AIEOP protocol (M. Schrappe, personal communication). It should be emphasized that truly low-risk patients can only be identified if the applied MRD technique is sufficiently sensitive (≤104).
MRD-based intermediate-risk patients
The group of MRD-based intermediate-risk patients (relapse rate of 20 to 30%) comprises ALL patients, who are MRD-positive at the end of induction treatment and are MRD-negative or positive at low levels (i.e., ≤104) before consolidation treatment.30,33 This group can have a substantial size and would need further treatment intensification, but this would imply that ∼75% of patients in this group are overtreated. The I-BFM-SG has shown that MRD information at later time points (e.g., at one year) has added value for the MRD-based intermediate-risk patients: MRD-positive patients (generally with low MRD levels) have a high chance of relapse, whereas MRD-negative patients have a low chance of relapse (P < 0.001 for T-ALL and P = 0.004 for precursor-B-ALL).33
The vast majority of relapses (∼90%) in the MRD-based intermediate-risk group occur after one year of treatment (see Figure 3A).30,33 Consequently, the MRD information at one year can potentially be used for treatment modification in MRD-positive intermediate-risk patients only.
Overall, this would imply that in up to 75% of ALL patients treatment might be adapted, based on MRD information at several follow-up time points during the first year of therapy.
MRD-based stratification of relapsed ALL patients
As suggested by the results of BFM study group,38 there is a subset of relapsed ALL patients with a good molecular response to re-induction chemotherapy. For such patients, BMT might not be required, which should be proven in a prospective treatment trial.
MRD-based stratification of ALL patients undergoing BMT
Based on available data,39– 42 MRD information should play an important role in the clinical BMT setting. It is generally accepted that BMT should be performed within the first half a year of remission, but patients with high MRD levels (≥ 10-3) at this time point are at very high risk of relapse post BMT. Future prospective trials should answer the question, whether further pre-transplant tumor load reduction can be achieved with intensified chemotherapy or post-transplant immunotherapy and whether this would improve post-BMT survival.
Choice of MRD Techniques
The three different MRD techniques differ in their sensitivity and applicability. Consequently, the choice of MRD techniques is dependent on the requirements of the clinical MRD study and, vice-versa, the applied MRD technique determines the possibilities for MRD-based treatment intervention (see Table 10).
If only high-risk patients need to be identified, GeneScanning of Ig/TCR gene rearrangements and flow cytometric MRD detection are well-suited (cheap and rapid) methods. If the clinical MRD study is focussing on the recognition of high-risk and low-risk patients within a specific subgroup of ALL, a sensitive MRD technique can be selected for that specific subgroup. For example, flow cytometry in case of a clinical MRD study in T-ALL or RT-PCR analysis of BCR-ABL fusion gene transcripts in case of a clinical MRD study focusing on Ph+ ALL.
Usage of different MRD techniques for different patients within the same treatment protocol should be avoided, unless full proof is provided that the obtained MRD results are fully identical, as has been demonstrated for RQ-PCR analysis of patient-specific Ig/TCR gene rearrangements and TEL-AML1 transcripts.48 Small single-center studies showed that results of MRD detection by flow cytometry and quantitative PCR of patient-specific Ig/TCR gene rearrangements are largely comparable.49,50 However, differences have been found in follow-up samples with low MRD levels (around 104), which are essential for recognition of low-risk patients.49,50 These discrepancies are probably caused by differences in cell sample processing, sensitivity, and expression of MRD results. So, harmonization of these topics is essential before MRD data obtained with different techniques become interchangeable.
Thus far, RQ-PCR detection of patient-specific Ig/TCR gene rearrangements is the most broadly applicable (> 90%) and sufficiently sensitive (≤ 104) MRD technique in ALL, which has proven to be realizable in multi-center clinical MRD studies.30,33,38 This probably explains why most European MRD studies in ALL use RQ-PCR analysis of Ig/TCR genes.
European Study Group on MRD Detection in ALL
Because of the high prognostic value of MRD information in ALL, clinical trials with MRD-based treatment intervention have been initiated or are currently being scheduled in most European countries. Thus far, these multicenter clinical MRD studies use PCR analysis of Ig/TCR genes as MRD technique. This European-wide implementation of MRD-based treatment protocols needs standardization, quality control, and rapid exchange of information between the PCR laboratories of the different clinical trials. Therefore, we recently initiated a unique European network of PCR laboratories to support the clinical ALL trials that aim at MRD-based treatment interaction.
The European Study Group on MRD Detection in ALL (ESG-MRD-ALL) consists of 23 experienced PCR laboratories from ten different European countries (NL, DE, FR, GB, AT, IT, ES, SE, DK, and CZ). The aims of the ESG-MRD-ALL are:
Standardization of the MRD-PCR techniques (particularly within each clinical protocol), including development of common guidelines for interpretation of MRD-PCR results.
Quality Control program with two quality control rounds per year.
Educational meetings twice per year, which include the evaluation of the quality control results.
Collaborative development and clinical implementation of new MRD strategies, e.g., introduction of new MRD-PCR targets.
Conclusions
Sensitive and quantitative MRD detection has proven to be clinically relevant in childhood ALL patients. Particularly the evaluation of early treatment response has high prognostic value, because this allows identification of low-risk and high-risk patients, which may profit from treatment reduction or treatment intensification, respectively. Also the pre-BMT MRD status has predictive value for post-BMT relapse-free survival. Comparable results are currently being obtained in adult ALL MRD studies, albeit that the relative sizes of the MRD-based risk groups differ from those in childhood ALL.
Although the prognostic value of MRD has been established, the reported MRD studies show remarkable differences in MRD-based risk group definition and the corresponding relapse-free survival rates. This might be related to the type of treatment protocol, the timing of the follow-up samples, or the applied MRD technique (particularly its sensitivity). Consequently, the precise prognostic value of MRD information has to be assessed carefully for each treatment protocol before MRD-based intervention can be implemented. Furthermore, standardization of MRD techniques and quality control of MRD results are essential for multicenter clinical MRD studies that aim for MRD-based treatment intervention.
Results of adult acute lymphoblastic leukemia (ALL) studies.
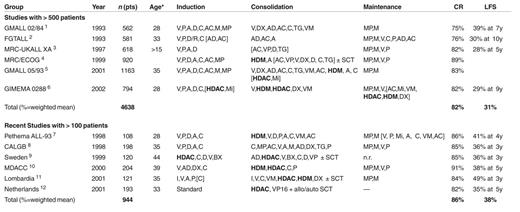
* median age or range if not available.
Abbreviations: n.r. not reported; CR complete remission; LFS, leukemia free survival; V, vincristine; P, prednisone; A, asparaginase; D, daunorubicine; C, cyclophosphamide; AC, cytarabine; M, methotrexate; MP, mercaptopurine; DX, dexamethasone; AD, adriamycin; TG, thioguanine; VM, teniposide; R, rubidazone; VP, etoposide; HDAC, high dose AC; HDM, high dose M; SCT, stem cell transplantation; Mi, mitoxantrone; BX, betamethasone; I, idarubicin.
New treatment and management options for adult acute lymphocytic leukemia (ALL).
| Molecular therapy |
| • Direct inhibition of molecular aberrations involved in pathogenesis |
| → tyrosine kinase inhibitor STI571, farnesyl transferase inhibitors |
| Antibody therapy |
| • Targeted suppression of leukemic blasts according to surface antigen expression |
| → see Table 5 |
| Non-myeloablative stem cell transplantation |
| • Utilization of graft-versus-leukemia effects |
| → Extension of stem cell transplantation indications to elderly/comorbid patients |
| Minimal residual disease evaluation |
| • Individual evaluation of treatment response |
| → Assessment of therapy elements, e.g., induction, novel therapeutics and risk stratification |
| Microarray analysis |
| • Analysis of gene expression profiles and selection of differentially expressed genes |
| → Identification of prognostic factors and target genes for novel therapeutics |
| Molecular therapy |
| • Direct inhibition of molecular aberrations involved in pathogenesis |
| → tyrosine kinase inhibitor STI571, farnesyl transferase inhibitors |
| Antibody therapy |
| • Targeted suppression of leukemic blasts according to surface antigen expression |
| → see Table 5 |
| Non-myeloablative stem cell transplantation |
| • Utilization of graft-versus-leukemia effects |
| → Extension of stem cell transplantation indications to elderly/comorbid patients |
| Minimal residual disease evaluation |
| • Individual evaluation of treatment response |
| → Assessment of therapy elements, e.g., induction, novel therapeutics and risk stratification |
| Microarray analysis |
| • Analysis of gene expression profiles and selection of differentially expressed genes |
| → Identification of prognostic factors and target genes for novel therapeutics |
Adverse prognostic factors in adult acute lymphocytic leukemia (ALL).*
| * Adverse prognostic factors as they emerged from the more than 3000 adult ALL patients treated in the GMALL (German Multicenter Studies for Adult ALL) trials.24,25 | |
| Clinical characteristics | Higher age: > 50 yrs, > 60 yrs |
| High WBC: > 300,00/μl in B-lineage | |
| Immunophenotype | Pro B (B-lin., CD10) |
| Early T (T-lin., CD1a, sCD3) | |
| Mature T (T-lin., CD1a, sCD3+) | |
| Cytogenetics/molecular genetics | t(9;22)/BCR-ABL or t(4;11)/ALL1-AF4 |
| Treatment response | Late achievement of CR: > 3, 4 weeks |
| MRD positivity | |
| * Adverse prognostic factors as they emerged from the more than 3000 adult ALL patients treated in the GMALL (German Multicenter Studies for Adult ALL) trials.24,25 | |
| Clinical characteristics | Higher age: > 50 yrs, > 60 yrs |
| High WBC: > 300,00/μl in B-lineage | |
| Immunophenotype | Pro B (B-lin., CD10) |
| Early T (T-lin., CD1a, sCD3) | |
| Mature T (T-lin., CD1a, sCD3+) | |
| Cytogenetics/molecular genetics | t(9;22)/BCR-ABL or t(4;11)/ALL1-AF4 |
| Treatment response | Late achievement of CR: > 3, 4 weeks |
| MRD positivity | |
Hematological and molecular complete response (CR) rates after induction in adult ALL.a
| Sensitivity | Hematologic CRb < 102 | Molecular CRc < 104 | Molecular CRd < 104 and negative |
|---|---|---|---|
| a Results of GMALL (German Multicenter Studies for Adult ALL) studies.33– 35 | |||
| b Morphological explanation | |||
| c Minimal residual disease (MRD) level below 104, positive or negative (minimum sensitivity 104) | |||
| d No detectable MRD at a sensitivity below 104 | |||
| Overall | 82% | ||
| Standard risk | |||
| B Lineage | 88% | 78% | 41% |
| T Lineage | 87% | 82% | 64% |
| High risk | 83% | ||
| Ph/BCR-ABL | 77% | 18% | 4% |
| Sensitivity | Hematologic CRb < 102 | Molecular CRc < 104 | Molecular CRd < 104 and negative |
|---|---|---|---|
| a Results of GMALL (German Multicenter Studies for Adult ALL) studies.33– 35 | |||
| b Morphological explanation | |||
| c Minimal residual disease (MRD) level below 104, positive or negative (minimum sensitivity 104) | |||
| d No detectable MRD at a sensitivity below 104 | |||
| Overall | 82% | ||
| Standard risk | |||
| B Lineage | 88% | 78% | 41% |
| T Lineage | 87% | 82% | 64% |
| High risk | 83% | ||
| Ph/BCR-ABL | 77% | 18% | 4% |
Antigen expression and treatment results with monoclonal antibodies in acute lymphocytic leukemia (ALL).
| Antigen | Expression on >20%a | Clinical Application Antibody | Stage | n | Tx | Results | Author |
|---|---|---|---|---|---|---|---|
| a Data from the GMALL (German Multicenter Studies for Adult ALL) central immunophenotyping, E.Thiel, S.Schwartz et al, Berlin, Germany. | |||||||
| CD19 | 95% B-precursor | AntiCD19+PAP | de novo child | 14 | CH | 43% CR d14 | Seibel et al 41 |
| 94% Mature B-ALL | 14 | AB+CH | 93% CR d14 | ||||
| AntiCD19+Ricin | de novo adult | 46 | AB+CH | No effect on MRD | Szatrowski et al 42 | ||
| AntiCD19+Genistein | ref/rel child | 15 | AB | 2 CR, 2 PR | Messinger et al 43 | ||
| CD20 | 41% B-precursor | Rituximab | de novo adult | 19 | AB+CH | 93% CR | Thomas et al 29 |
| 86% Mature B-ALL | 86% OS 1y | ||||||
| CD52 | 66% B-/T-lineage | Campath | ref/rel. adult | 5 | AB | 0 CR, 0 PR | Faderl et al 44 |
| Antigen | Expression on >20%a | Clinical Application Antibody | Stage | n | Tx | Results | Author |
|---|---|---|---|---|---|---|---|
| a Data from the GMALL (German Multicenter Studies for Adult ALL) central immunophenotyping, E.Thiel, S.Schwartz et al, Berlin, Germany. | |||||||
| CD19 | 95% B-precursor | AntiCD19+PAP | de novo child | 14 | CH | 43% CR d14 | Seibel et al 41 |
| 94% Mature B-ALL | 14 | AB+CH | 93% CR d14 | ||||
| AntiCD19+Ricin | de novo adult | 46 | AB+CH | No effect on MRD | Szatrowski et al 42 | ||
| AntiCD19+Genistein | ref/rel child | 15 | AB | 2 CR, 2 PR | Messinger et al 43 | ||
| CD20 | 41% B-precursor | Rituximab | de novo adult | 19 | AB+CH | 93% CR | Thomas et al 29 |
| 86% Mature B-ALL | 86% OS 1y | ||||||
| CD52 | 66% B-/T-lineage | Campath | ref/rel. adult | 5 | AB | 0 CR, 0 PR | Faderl et al 44 |
Late effects of cranial irradiation.
| • Mineralizing microangiopathy and leukoencephalopathy leading to seizure, deficits in intellectual function, memory loss and learning difficulty |
| • Second cancers including malignant brain tumor, meningioma, parotid gland carcinoma, thyroid carcinoma, basal cell carcinoma, and (rarely) sarcoma |
| • Endocrinopathy resulting in growth retardation, obesity, and osteoporosis |
| • Craniofacial deformities |
| • Mineralizing microangiopathy and leukoencephalopathy leading to seizure, deficits in intellectual function, memory loss and learning difficulty |
| • Second cancers including malignant brain tumor, meningioma, parotid gland carcinoma, thyroid carcinoma, basal cell carcinoma, and (rarely) sarcoma |
| • Endocrinopathy resulting in growth retardation, obesity, and osteoporosis |
| • Craniofacial deformities |
Characteristics of the techniques currently employed for minimal residual disease (MRD) detection in acute lymphoblastic leukemia (ALL).
| Flow Cytometric Immunophenotyping | PCR Analysis of Chromosome Aberrations (mainly detection of fusion gene transcripts) | PCR Analysis of Ig/TCR Genes (junctional region specific approach) | |
|---|---|---|---|
| * In childhood ALL this particularly concerns t(12;21)(TEL-AML1) and in adult ALL particularly t(9;22)(BCR-ABL). | |||
| ** This mainly concerns del(1)(p32 p32) with SIL-TAL1 fusion and t(5;14) with aberrant HOX11L2 expression, together occurring in 25-35% of childhood T-ALL and in 15-20% of adult ALL.11,12 | |||
| Sensitivity | 10–3–10–4 | 10–4–10–6 | 10–4–10–5 |
| Applicability | |||
| Precursor-B-ALL | 60-98% | 40-45%* | 90-95% |
| T-ALL | 90-95% | 15-35%** | 90-95% |
| Advantages |
|
|
|
| Disadvantages |
|
|
|
| Flow Cytometric Immunophenotyping | PCR Analysis of Chromosome Aberrations (mainly detection of fusion gene transcripts) | PCR Analysis of Ig/TCR Genes (junctional region specific approach) | |
|---|---|---|---|
| * In childhood ALL this particularly concerns t(12;21)(TEL-AML1) and in adult ALL particularly t(9;22)(BCR-ABL). | |||
| ** This mainly concerns del(1)(p32 p32) with SIL-TAL1 fusion and t(5;14) with aberrant HOX11L2 expression, together occurring in 25-35% of childhood T-ALL and in 15-20% of adult ALL.11,12 | |||
| Sensitivity | 10–3–10–4 | 10–4–10–6 | 10–4–10–5 |
| Applicability | |||
| Precursor-B-ALL | 60-98% | 40-45%* | 90-95% |
| T-ALL | 90-95% | 15-35%** | 90-95% |
| Advantages |
|
|
|
| Disadvantages |
|
|
|
Chromosome aberrations in acute lymphoblastic leukemia (ALL) as (reverse transcriptase) polymerase chain reaction [(RT-)PCR] targets for minimal residual disease (MRD) detection.a
![Chromosome aberrations in acute lymphoblastic leukemia (ALL) as (reverse transcriptase) polymerase chain reaction [(RT-)PCR] targets for minimal residual disease (MRD) detection.a](https://ash.silverchair-cdn.com/ash/content_public/journal/hematology/2002/1/10.1182_asheducation-2002.1.162/3/m_179hemtable8.jpeg?Expires=1767710372&Signature=3cSYB-mwD8aTzETKrpOleoXf9Fus4oEq0KWPcQ0QRV~myrU14ICMoitsrPOxLpFT8qIFLIZEwxCXoW0PjnAI~hkHIddxMY2zOGehgZBEmnwVW5REbZvVMhyrqin2Mpj~i3HoomfJs0LJgOLcx6LizuoUe2xwvzxEzEpXL2vOotsbPBS5iAD9ybZyNmWE0-umaS8fZi8dEGO9~fO3S-UrroVQVMTMNKpnkBG1bfu9RbTTjA~WvNZ7LY5m3k9v7SUZXKroAjX7E25VK8Q7hb9fFUPVhWWLs51x7yZkZQbHo1ItoDUTQq0WKn2eN~XZWQceTOQ3p2UwFSHtZrMmfwEK8w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
a The detection limit of PCR analysis of chromosome aberrations is 10–4 to 10–6.
b The indicated percentages represent frequencies within the precursor-B-ALL and T-ALL groups.
c In infant ALL, the frequency of t(4;11) can be as high as 50% and the total group of 11q23 aberrations as high as 70%.
Frequencies and stability of minimal residual disease polymerase chain reaction (MRD-PCR) targets in childhood precursor-B-cell acute lymphoblastic leukemia.
| Stability at Relapse** | |||||
|---|---|---|---|---|---|
| PCR Target | Frequency at Diagnosis* | Monoclonality at Diagnosis* | Monoclonal | Oligoclonal | Total |
| * see references 15-17. | |||||
| ** see reference 20. | |||||
| Abbreviations: NT, not tested. | |||||
| IGH | 95% | 60-70% | 85% | 44% | 64% |
| Vh-Jh | 95% | 88% | 47% | 69% | |
| Dh-Jh | 20% | 57% | 38% | 43% | |
| IGK-Kde | 50% | 90% | 95% | 40% | 90% |
| Vκ-Kde | 45% | 95% | 40% | 91% | |
| intron-Kde | 25% | 86% | 0 | 87% | |
| TCRG (Vγ-Jγ) | 55% | 60-65% | NT | NT | 75% |
| TCRD | 40% | 60% | 86% | 26% | 63% |
| Vδ2-Dδ3 | 35% | 81% | 31% | 63% | |
| Dδ2-Dδ3 | 7% | 100% | 14% | 63% | |
| Stability at Relapse** | |||||
|---|---|---|---|---|---|
| PCR Target | Frequency at Diagnosis* | Monoclonality at Diagnosis* | Monoclonal | Oligoclonal | Total |
| * see references 15-17. | |||||
| ** see reference 20. | |||||
| Abbreviations: NT, not tested. | |||||
| IGH | 95% | 60-70% | 85% | 44% | 64% |
| Vh-Jh | 95% | 88% | 47% | 69% | |
| Dh-Jh | 20% | 57% | 38% | 43% | |
| IGK-Kde | 50% | 90% | 95% | 40% | 90% |
| Vκ-Kde | 45% | 95% | 40% | 91% | |
| intron-Kde | 25% | 86% | 0 | 87% | |
| TCRG (Vγ-Jγ) | 55% | 60-65% | NT | NT | 75% |
| TCRD | 40% | 60% | 86% | 26% | 63% |
| Vδ2-Dδ3 | 35% | 81% | 31% | 63% | |
| Dδ2-Dδ3 | 7% | 100% | 14% | 63% | |
Comparison between large prospective minimal residual disease (MRD) studies in pediatric acute lymphocytic leukemia (ALL).*
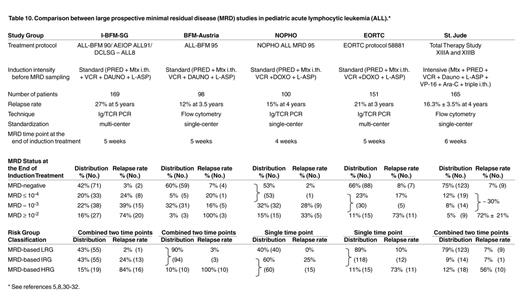
* See references 5,8,30-32.

Hypothetical graph showing the kinetics of leukemic cell decrease and re-growth in several ALL patients during and after treatment with the I-BFM-SG treatment protocol.
MRD curves represent individual patients of the three MRD-based risk groupstwo patients with slow MRD clearance (high-risk group), two patients with moderate MRD clearance (intermediate-risk group), and one patient with rapid MRD clearance (low-risk group).30 The detection limit of cytomorphologic techniques as well as the detection limit of flow cytometric immunophenotyping and PCR techniques are indicated. I: induction treatment; C: consolidation treatment; II: re-induction treatment.
Hypothetical graph showing the kinetics of leukemic cell decrease and re-growth in several ALL patients during and after treatment with the I-BFM-SG treatment protocol.
MRD curves represent individual patients of the three MRD-based risk groupstwo patients with slow MRD clearance (high-risk group), two patients with moderate MRD clearance (intermediate-risk group), and one patient with rapid MRD clearance (low-risk group).30 The detection limit of cytomorphologic techniques as well as the detection limit of flow cytometric immunophenotyping and PCR techniques are indicated. I: induction treatment; C: consolidation treatment; II: re-induction treatment.

Consecutive rearrangements in theIGKlocus, resulting in the two main types of Kde rearrangements.
Most IGK gene rearrangements start with a Vκ-Jκ rearrangement. The functionality of this rearrangement can be disrupted by rearrangement of Kde (kappa deleting element), which deletes the Cκ segment region when the intron recombination signal sequence (intron RSS) is used or deletes the Vκ-Jκ-Cκ region when Kde rearranges to a Vκ gene segment. Both types of Kde rearrangements will delete the IGK gene enhancers (iEκ and 3IEκ), which probably precludes further rearrangements in the IGK locus.
Consecutive rearrangements in theIGKlocus, resulting in the two main types of Kde rearrangements.
Most IGK gene rearrangements start with a Vκ-Jκ rearrangement. The functionality of this rearrangement can be disrupted by rearrangement of Kde (kappa deleting element), which deletes the Cκ segment region when the intron recombination signal sequence (intron RSS) is used or deletes the Vκ-Jκ-Cκ region when Kde rearranges to a Vκ gene segment. Both types of Kde rearrangements will delete the IGK gene enhancers (iEκ and 3IEκ), which probably precludes further rearrangements in the IGK locus.

(A) Relapse-free survival of the three MRD-based risk groups of children treated for ALL according to protocols of the International BFM Study Group. The three risk groups were defined by combined MRD information at the end of induction treatment and before consolidation treatment (see30for details).
(B) Relapse-free survival according to the qualitative (presence or absence) and quantitative detection of MRD after the completion of induction therapy in EORTC trial 58881 (with courtesy to Dr. H. Cavé and Prof. E. Vilmer).32,36
(A) Relapse-free survival of the three MRD-based risk groups of children treated for ALL according to protocols of the International BFM Study Group. The three risk groups were defined by combined MRD information at the end of induction treatment and before consolidation treatment (see30for details).
(B) Relapse-free survival according to the qualitative (presence or absence) and quantitative detection of MRD after the completion of induction therapy in EORTC trial 58881 (with courtesy to Dr. H. Cavé and Prof. E. Vilmer).32,36

(A) Probability of event-free survival according to the MRD levels after the second block of the ALL-REZ BFM treatment protocols (with courtesy to Dr. C. Eckert and Prof. G. Henze).38(B) Probability of event-free survival after allogeneic BMT according to the MRD levels in pre-BMT BM samples (with courtesy to Dr. P. Bader).42
(A) Probability of event-free survival according to the MRD levels after the second block of the ALL-REZ BFM treatment protocols (with courtesy to Dr. C. Eckert and Prof. G. Henze).38(B) Probability of event-free survival after allogeneic BMT according to the MRD levels in pre-BMT BM samples (with courtesy to Dr. P. Bader).42
