
Abstract
Significant advancements in the care of patients with chronic lymphocytic leukemia (CLL) have occurred over the past decade. Nonetheless, CLL remains incurable outside of allogeneic transplantation. CLL is the most common leukemia in the United States and Europe, and new treatments and therapeutic strategies are clearly needed. To address this need, the pathogenesis of CLL has been an area of intense ongoing investigation. These international efforts illuminate a complex biology that is reliant on the interplay of inherited, environmental, and host factors. This broad review will discuss the recent advances in our understanding of CLL biology including the elucidation of inherited and acquired genetic changes; the role of the B-cell receptor and B-cell receptor signaling; CLL cell kinetics; and the interactions in the microenvironment between CLL cells, other immune cells, and stromal elements. This improved understanding of disease pathogenesis is facilitating the development of novel therapeutic treatment strategies.
Chronic lymphocytic leukemia (CLL) remains an enigmatic disease. Although the first clinical description of chronic lymphoid leukemia was published over 150 years ago, the etiology of CLL is unknown, the cell of origin of CLL is unknown, and CLL remains incurable outside of allogeneic transplantation. CLL is the most common leukemia in the United States, with approximately 15,500 new diagnoses per year. Because of the relatively long survival of patients with CLL, it is by far the most prevalent leukemia, with an estimated 95,000 Americans living with CLL.1 CLL lymphocytes have a characteristic immunophenotype: CD5+, CD19+, CD20dim, CD23+, and surface immunoglobulindim. Although the majority of patients are asymptomatic at diagnosis, the relentless accumulation of CLL lymphocytes leads to symptomatic disease, need for CLL-directed therapy, disease-related complications, and approximately 4500 CLL attributable deaths per year in the United States.
Several characteristics of CLL facilitate basic and translational research: (i) the high population prevalence; (ii) the malignant cells are easily obtained through venous phlebotomy; (iii) most patients have an asymptomatic phase that allows for longitudinal evaluation; and (iv) CLL is has a relatively long disease-specific survival. Therefore, CLL has become a model system for the investigation of B-cell lymphoproliferative disorders. In the 5 years since the biology of CLL was last broadly reviewed in the American Society of Hematology education session, tremendous progress has been made in the understanding of CLL disease biology, and this review will focus on these discoveries. Specifically, important advances have been made in identifying inherited and acquired genetic mutations, the role of B-cell receptor (BCR) signaling, and the interplay between the malignant B cells and the tumor microenvironment. These advances reveal CLL to be a disease that is dependent on on the interplay of inherited, environmental, and host factors.
Inherited Genetic Factors
The most important risk factor for the development of CLL is a family history of CLL. Among patients with newly diagnosed CLL, 8% to 10% have a family history of CLL. CLL has a heritability that is twice that of common solid tumors with known low-prevalence, high penetrance causative genes, such as breast and colon cancer.2,3 Pedigree analysis using data from the Swedish Family Cancer Database showed the relative risk of CLL among first-degree relatives of persons with CLL to range between 7.0 and 8.5. This equivalent risk across all first-degree relatives suggests an inherited, rather than environmental, basis for familial CLL.4 Though families with four or more cases of CLL have been reported, they are extremely rare. Some CLL kindreds suggest a dominant inheritance, therefore, genome-wide linkage studies in familial CLL have been performed. The largest and most recent reported study added 101 new cases of familial CLL to a previously reported cohort of 105 families.5 This study was the first statistically significant genome-wide linkage scan and identified chromosome 2q21.2 as associated with inheritance of CLL. However, no causative genes have been identified at this locus. Overall, linkage studies in CLL have been limited by the low number of affected individuals per family, late age of onset, and presumed genetic heterogeneity.
Complete sequencing of the human genome revealed large numbers of common single nucleotide polymorphisms (SNPs). This discovery led to the hypothesis that the inheritance of complex diseases may be due to the coinheritance of these common variants. Di Bernardo et al6 performed a multistage genome-wide SNP association (GWA) study to evaluate the contribution of common variants to the inheritance of CLL. This study identified six novel loci associated with the development of CLL, thus providing the first evidence for the contribution of common (population prevalence > 5%) genetic variants to the development of CLL. Five of these SNPs have been validated in an independent GWA.7 Subsequently, four additional susceptibility loci have been identified through follow-up analyses from the initial Di Bernardo study.8 Identification of these genes informs our understanding of CLL biology. For example, the GWA linked interferon regulatory factor 4 (IRF4), a key regulator of lymphocyte maturation and proliferation, with risk of developing CLL. The 10 loci identified to date individually confer a small risk of disease, with relative risks ranging from 1.2 to 1.6. As a group, these risk alleles account for < 10% of the total heritability of CLL. Because GWA can detect only those alleles with a population prevalence of > 5%, it is possible that much of the inherited risk of CLL is due to uncommon inherited variants (prevalence < 5%). Massively parallel sequencing technologies may enable the discovery of these rare variants and illuminate the “hidden heritability” in CLL.
Acquired Genetic Factors
In a landmark report by the German CLL Study Group, Dohner et al9 showed that acquired chromosomal abnormalities involving chromosomes 11, 12, 13, and 17 are common in CLL, and that these abnormalities predict both time to first treatment and CLL-specific survival. The genes principally responsible for the adverse prognosis associated with del 17p13 and del 11q22 were recognized to be TP53 and ATM, respectively. The genes that contribute to CLL pathogenesis in trisomy 12 remain unknown. There is ongoing debate regarding whether trisomy 12 confers an increased risk of disease progression; unlike the Dohner study, our institutional experience is that patients with trisomy 12 do follow a somewhat more aggressive disease course than those patients with normal FISH (fluorescence in situ hybridization).10 Deletion of 13q14 is the most common and most favorable cytogenetic abnormality in CLL. The responsible genes at this locus were initially unclear. In 2002, Calin et al11 showed that the microRNA 15/16 (miR 15/16) cluster were the critical deleted genes in this region. MicroRNAs are small (approximately 20 nucleotides) nonprotein coding RNAs that modulate the level of specific proteins by binding sequence-complimentary mRNAs. miR-15 and miR-16 were subsequently shown to be negative regulators of BCL2.12 This discovery is notable in that it was the first association of clinical disease with microRNAs.
The Dohner study9 established interphase cytogenetics (FISH) as the clinical standard of care for evaluation of chromosomal aberrations in CLL because the low proliferative capacity of most CLL limits the clinical utility of metaphase cytogenetics. However, emerging data shows that the evaluation of four (or five) chromosomal loci by FISH obscures the significant heterogeneity and complexity of acquired chromosomal defects in CLL. The application of high-density SNP arrays illuminates some of this complexity. For example, copy number neutral loss of heterozygosity is observed in a significant number of CLL cases, and this finding cannot be detected with either FISH or standard karyotyping. Using SNP arrays, Ouilette et al13 showed significant heterogeneity among 13q14 deletions, with some deletions extending centromeric to include the retinoblastoma gene. A follow-up study showed that 13q14 deletions that include retinoblastoma are associated with genomic complexity,14 a finding associated with aggressive disease in prior clinical reports. Although data currently available are inadequate to support the widespread application of SNP arrays to routine clinical care, this technology may ultimately be a useful adjunct to routine clinical FISH.
At the chromosomal level, del 17p13 is more homogenous than other acquired genomic defects. High-density SNP arrays showed that the deletion breakpoint typically falls within chromosomal band 17p11.2 and extends telomeric to include most of the p arm.15 Deletions of 17p13 are almost always monoallelic, and it was initially unclear why monoallelic loss of TP53 would cause such a dramatic change in CLL proliferative capacity, chemosensitivity, and prognosis. It is now understood that > 80% of cases with del 17p13 have single nucleotide somatic mutations of the TP53 allele on the other chromosome 17 (the allele in trans), thereby causing near complete loss of TP53 function. Although uncommon, TP53 inactivating mutations can occur in the absence of del 17p13, and these mutations apparently confer an equivalent adverse risk as del 17p13.16 TP53 is a multimeric protein with four identical subunits, and these inactivating mutations may act in a dominant negative manner, thus abrogating TP53 function even in the presence of a wild-type allele in trans. Although only 5% of CLL cases show deletion or mutation of 17p13 at diagnosis, somatic mutations occur in approximately one-third of relapsed or chemotherapy refractory patients. Whether this represents clonal selection or clonal evolution or both is an area of active investigation.
Epigenetic changes are also relevant to the pathogenesis of CLL. Epigenetic changes are noninherited chromosomal modifications that affect gene transcription, such as methylation of gene promoters and acetylation of histone-bound DNA. An example of epigenetic change, global hypomethylation of DNA, has been described in CLL. Between 2% and 8% of CpG islands (gene promoter elements) are aberrantly methylated in CLL when compared with normal B cells, a finding that suggests DNA methylation broadly affects the transcriptional profile of CLL. A germline mutation in death-associated protein kinase 1 (DAPK1) that segregated with risk of CLL was identified by Raval et al18 in a family with multiple CLL cases. The authors showed that DAPK1 expression is silenced through promoter methylation in the majority of CLL cases, suggesting a central role for both epigenetic modification and DAPK1 in CLL leukemogenesis.18 Using high-density methylation microarrays evaluating over 27,000 CpG sites, Kanduri et al19 identified differential patterns of methylation that were dependent on the mutation status of the BCR. Poor-risk CLL showed methylation profiles that facilitated signaling through proliferative cellular pathways, including MAPK (mitogen-activated protein kinase) and NF-κB (nuclear factor-κ light-chain enhancer of activated B cells), a finding that directly relates the clinical phenotype of poor-risk CLL to methylation of BCR-mediated signaling pathways.
The Role of BCR in CLL
Investigation of the immunoglobulin gene has led to findings that are central to the current understanding of CLL (Table 1). During B-cell development, variable-diversity-joining (VDJ) recombination generates an immunoglobulin heavy chain (IGH), and an immunoglobulin light chain (IGL) is created through isotype-specific VJ recombination. There is tremendous combinatorial potential in this process, with perhaps 1010 potential IGH-IGL sequence combinations. However, patients with CLL use a highly restricted set of BCRs.20 Approximately 14% of all CLL cases express the heavy chain VH1–69, and an additional 18% express VH4–34. This finding of marked restriction of the IGH repertoire led to the hypothesis that tonic stimulation by specific auto or alloantigens drives expansion of the malignant clone.21,22 Intraclonal diversification of some CLL further supports the antigen-drive hypothesis.23
The mutation status of the immunoglobulin heavy chain (IGVH) as a central determinant of CLL biology
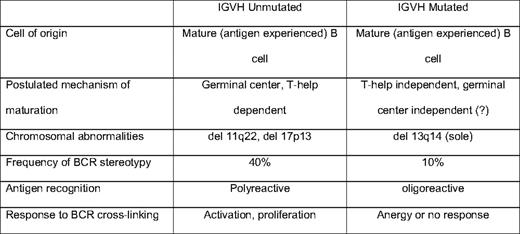
During the germinal center reaction, somatic mutations are generated in the BCR to increase affinity for target antigen. Approximately half of CLL cases show IGH mutations, and the other half show an “unmutated” IGH, typically defined as < 2% deviation from germline sequence. Patients with mutated IGH typically follow a more indolent disease course than those with unmutated IGH24 . This initially led to the hypothesis that CLL may be two distinct entities, one deriving from naive (IGH unmutated) B cells and the other from postgerminal center memory (IGH mutated) B cells. However, gene expression profiling studies in CLL show both IGH mutated and unmutated CLL to be derived from memory B cells.25,26 The mechanism by which some CLL escape IGH mutation is unclear. One hypothesis proposes that certain antigens favor a helper T-cell independent or germinal center independent maturation that does not involve IGH mutation. Alternatively, the IGH unmutated CLL cells may be autoreactive and targeted for apoptosis, but avoid cell death through either tonic BCR stimulation or transforming genetic events.
Recent experiments using phage display libraries have attempted to better define the types of antigens and epitopes recognized by IGH mutated and unmutated CLL. Monoclonal antibodies (MAbs) generated from the BCR of IGH-mutated CLL bound epitopes with structurally related amino acid motifs, whereas unmutated CLL recognized multiple different epitopes.27 This suggests that in vivo multiple, potentially structurally divergent antigens can bind and stimulate CLL cells through the BCR. This conclusion is provocative because six of eight of the CLL MAbs investigated had “stereotyped” BCRs. Stereotypy is the observation of near complete sequence homology of the complimentarity-determining region 3 (CDR3) of the IGH28 ; the CDR3 largely defines the antigen specificity of the BCR. For example, approximately 1% of all CLL express VH1–69 that are virtually identical within the CDR3.29 The presence of a stereotyped BCR is associated with adverse risk, independent of IGH mutation status, although stereotypy is more commonly observed among unmutated (40%) than mutated CLL (10%). The observation of stereotypy has been forwarded as evidence of selective antigenic pressure in CLL; however, MAbs derived from these BCRs may be oligo- or polyreactive in vivo. MAbs derived from stereotyped BCRs have also been shown to react with conserved epitopes exposed on apoptotic cells.30 The selection of stereotyped BCRs may be related to a combination of antigenic pressure and CDR3 sequence facilitated BCR signaling.
Given the clear importance of the BCR in the clinical behavior of CLL, the role of BCR-mediated signaling is another area of significant interest. The surface density of the BCR is lower in CLL than in most B-cell subsets, suggesting CLL may have attenuated signaling responses to antigen binding. Experimental evidence shows that, in vitro, approximately half of all CLL retain BCR-mediated signaling, and that the IGH mutation status and other molecular characteristics used for clinical prognostication (eg, CD38 and ZAP70 [zeta-associated protein 70]) are important determinants of BCR responsiveness. After cross-linking the BCRs with isotype-specific antibodies, Lanham et al31 showed global increases in tyrosine phosphorylation, a marker of induction of intracellular signaling. The responsiveness to cross-linking was strongly associated with IGH mutation status (P < .0005), as well as expression of CD38 (P < .05).31 Using gene expression profiling, Guarini et al32 showed that BCR cross-linking with anti-IgM led to the transcription of gene pathways regulating cell-cycle regulation, cytoskeletal organization, and proliferation; but, these changes were only observed among IGH unmutated cases. BCR ligation leads to enhanced intracellular signaling among ZAP70 expressing CLL, and the introduction of ZAP70 into CLL not otherwise expressing ZAP70 augments BCR-mediated signaling, suggesting a direct role for ZAP70 in BCR-mediated signaling.33 Muzio et al34 showed that CLL B cells that do not respond to BCR ligation (typically IGH-mutated cases) show activation of cellular pathways that suggest anergy. BCR signaling in vivo is likely even more complicated: assuming that there are multiple antigens capable of binding the BCR, the binding affinity for a specific antigen likely determines whether the response is activating or anergic. Although this complexity in vivo remains incompletely understood, in general, BCR ligation in IGH unmutated CLL leads to predominantly activating and proliferative responses, whereas BCR signaling in IGH-mutated CLL favors anergic and antiapoptotic responses. Additionally, the adverse risk associated with ZAP70 and CD38 expression may be directly related to their effects on cell signaling and activation.
CLL Cell Kinetics
The conventional view of CLL had been that it is primarily a disease of failed apoptosis and passive accumulation. This view is supported by the observation that the great majority (> 98%) of peripherally circulating CLL cells are arrested in G0 or the early G1 phase and have overexpression of antiapoptotic proteins, such as BCL2. Other observations suggest that CLL may have significant proliferative capacity. As discussed in the prior section, IGH unmutated CLL has the capacity for proliferative responses to BCR ligation. Many CLL express markers of cellular activation (eg, CD38, CD49d, and CD69), providing a link between CLL cell biology and prognostics. Clonal evolution and Richter's transformation are observed in some cases. In a series of elegant experiments, Messmer et al 35 evaluated the in vivo kinetics of CLL by having patients consume fixed doses of deuterated “heavy” water (2H2O) for 84 days. The deuterium was incorporated into newly synthesized DNA during the S phase. Using mass spectrometry, the “birth” and “death” rates of CLL cells were then determined. Although there was significant variability in birth rates (0.11%–1.76%/day), all patients had a rate of new cell formation of at least 109 new CLL cells per day. Patients with higher birth rates (exceeding 0.35%/day) were more likely to have symptomatic disease. Significant variability was also observed in the cellular death rate, and the balance between birth and death likely determines both white blood cell trend and risk of progression.35
The deuterated water experiments also provided novel insights into CLL intraclonal heterogeneity and CLL cell trafficking. By flow sorting the CD38+ and CD38– fractions from CLL patients receiving deuterated water, Calissano et al36 showed that the CD38+ fraction proliferated at a greater rate than the CD38– fraction. It has previously been shown that, regardless of the proportion of CD38+ cells, these cells show significantly increased expression of proliferation factors ZAP70, Ki67, and telomerase when compared with the CD38– fraction.37 In a more recent report, Calissano et al provided preliminary evidence that “new” CLL cells have a distinct immunophenotype: CD5hiCXCR4low, whereas “resting” CLL cells are CD5lowCXCR4hi. Gene expression array analysis of flow sorted “new” and “resting” cell fractions that showed that “new” cells differentially express genes related to proliferation, cellular activation, and cell signaling, whereas “resting” cells predominantly express genes involved in apoptosis, cell death, and migration.38 These studies have led to the hypothesis that CLL cells in the blood compartment eventually enter a pathway of cellular senescence. Upregulation of CXCR4 (a CXC chemokine receptor) promotes reentry into the tumor microenvironment, where the cells receive prosurvival signals. Rather than undergoing apoptosis, a subset of “resting” CLL cells are activated, proliferate, downregulate CXCR4, and are then released back into the peripheral blood. These studies shed light on the phenotypic continuum of CLL cells and better define the life cycle of a CLL B cell, as well as the central role of the tumor microenvironment in maintaining the malignant clone.
The Tumor Microenvironment in CLL
Over the past decade, the essential role of the tumor microenvironment in the survival and progression of CLL has become increasingly clear. The tumor microenvironment describes an admixture of malignant cells with host immune cells, stromal elements, and vascular cells that create a niche wherein signals can be transmitted through antigen presentation, cell–cell interactions, and paracrine signaling. The microenvironments differ in the bone marrow and secondary lymphoid organs, where the former contains mesenchymal stromal cells (MSC) within “vascular niches” and in the latter nurse-like cells (NLCs), T cells and follicular dendritic cells are present.
NLCs are large CD14+ mononuclear cells that are abundant in the secondary lymphoid organs of patients with CLL.39 NLCs and MSCs constitutively express the chemokines CXCL12 (stromal-derived factor-1α, Sdf-1) and CXCL13. CLL cells express CXCR4 (CD184), the receptor for Sdf-1, and in vitro studies have shown that engagement of Sdf-1 with CXCR4 promotes cell survival through activation of the signal transducer and activator of transcription 3 (STAT3) and MAPK pathways. Blockade of this interaction with a small molecule inhibitor leads to decreased CLL viability, suggesting this interaction is a potential therapeutic target.40 Additional studies have shown that CLL cell viability can be significantly enhanced in vitro with Sdf-1, although viability is further enhanced with NLCs over Sdf-1, suggesting that NLCs also deliver additional prosurvival signals. It is now understood that, in addition to Sdf-1, NLCs also secrete B-cell activating factor (BAFF) and a proliferation-inducing ligand (APRIL). Engagement of these ligands with CLL cells induces intracellular NF-κB1 and MCL-1, whereas sdf-1 induces ERK1/2 and AKT. As such, NLCs can deliver multiple prosurvival signals that utilize distinct signaling pathways.41 Engagement of CD40, expressed on the surface of CLL cells, with CD154 (CD40L), expressed on CD4+ T cells found in CLL pseudofollicles, also induces the NF-κB pathway. These diverse interactions have important implications for CLL therapy, particularly as mechanisms of chemotherapy resistance. A recent report showed that in vitro coculture of CLL cells with CD154 expressing fibroblasts induced 1000-fold resistance to ABT-737, a small molecular inhibitor of BCL2 and BCL-XL.42 Similarly, coculture of CLL cells with MSCs induces resistance to fludarabine, cyclophosphamide, and dexamethasone through maintenance of MCL-1.43 Taken together, these in vitro data strongly suggest that the tumor microenvironment enhances CLL cell resistance to both apoptosis and chemotherapy.
Investigation of novel mechanisms of interaction between the tumor microenvironment and CLL cells is an active area of research. CLL cells have the capacity to affect local angiogenesis within the vascular niche. Microvessel density is increased in bone marrow of CLL patients, with highest densities found at the periphery of lymphoid aggregates. CLL cells have the capacity to secrete a variety of angiogenic cytokines, including vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), and thrombospondin-1 (TSP-1).44 More recently, Ghosh et al45 described the contribution of CLL cell-derived microvesicles as a mechanism by which CLL cells can module the local microenvironment. Microvesicles are cell membrane-derived particles that can deliver cell surface receptors, activated signaling proteins, or nucleic acids to target cells. The authors found that CLL cell-derived microvesicles were increased in patients with advanced-stage CLL, and that microvesicles could activate AKT/mTOR signaling in MSCs.45 Thus, microvesicles represent a novel mechanism of CLL signaling within the microenvironment. Finally, toll-like receptors (TLRs) are cell surface receptors that are part of the innate immune system that bind structurally conserved microbial antigens and activate immune responses. Recent data shows that TLRs are expressed and functional in CLL; binding of TLRs with cognate antigen-activated NF-κB expression and induced surface expression of activation markers CD25 and CD80.46 Given the architectural complexity, number of different cell types present, and clinical importance of these interactions, investigation of the tumor microenvironment will continue to be an active and fruitful area of research in CLL.
T-Cell Abnormalities in CLL
Most malignancies are associated with decreased numbers of circulating T cells, but in CLL they are elevated 2.5 to 4 times normal, at least pretherapy.47 Although T cells are increased in number, the T-cell repertoire is significantly contracted in CLL, with oligoclonal and monoclonal subsets.48,49 CLL cells secrete immunomodulatory cytokines, such as interleukin-6, interleukin-10, and tumor growth factor-β (TGF-β), which shift the helper T-cell response from a Th1 response to an anergic Th2 response.50,51 Suppressive regulatory T cells (Treg) are also increased in patients with CLL.52 A mechanism for this increase may be direct CLL–T-cell contact through CD27 and CD70; this interaction also induces BCL2 within the Treg, making the Treg cell population in CLL patients more resistant to apoptosis than in normal individuals.53 These qualitative and quantitative T-cell abnormalities likely allow the CLL lymphocytes to avoid cell-mediated immune responses, despite the fact that CLL cells express tumor-specific antigens that can be presented by major histocompatibility complex molecules.
To better understand the mechanism underlying impaired T-cell responses in CLL, Gorgun et al54 performed gene expression array analyses of purified CD4+ and CD8+ T cells. Among CD4+ T cells, genes related to cell differentiation were differentially expressed between T cells derived from previously untreated CLL patients and healthy controls. Gene pathways controlling cytoskeleton formation, vesicle trafficking, and cytotoxicity were differentially expressed in CD8+ T cells. When CLL cells were cocultured with CD4+ or CD8+ T cells obtained from healthy controls, the abnormal patterns of gene expression were induced, suggesting that these T-cell defects are due to direct cell–cell contact.54 In light of the observed dysregulation of cytoskeleton assembly genes, a subsequent study by Ramsay et al55 evaluated the capacity of T cells to form a normal immunological synapse. This study showed that T cells derived from CLL patients could not form normal cell–cell contact and that recruitment of T-cell signaling proteins to the synapse was blocked. Similarly, coculture of CLL cells with donor T cells blocked the formation of an immunologic synapse.55 The inability of T cells derived from CLL patients to form a normal immunologic synapse likely impairs both antigen recognition and cellular cytotoxicity. Taken together, this global impairment in T-cell function may be an important cause of tumor progression, increased susceptibility to infection, and secondary malignancies in patients with CLL.
Using Novel Biologic Insights to Develop an Integrated Model of Disease Biology
Detailed laboratory-based and translational investigation has yielded tremendous progress in our understanding of CLL. One of the significant challenges at this time is to assemble this body of knowledge into a unified model of disease pathogenesis. Epidemiologic data implies the importance of ethnicity (likely a marker of genetic risk) and age (a marker of immune senescence). A proposed model follows: in a genetically susceptible host, tonic stimulation by a stereotyped antigen—likely a low-affinity autoantigen—initiates expansion of a premalignant clone. Through some combination of tonic antigenic stimulation, acquisition of somatic genetic mutations, and timely support from the tumor microenvironment, the potentially autoreactive clone escapes immune surveillance and begins to expand. In the tumor microenvironment, cells receive proliferative signals from CD4+ T cells and antiapoptotic signals from NLCs and MSCs. Initially supported by the tumor microenvironment, the increasing clone may begin to further modulate the microenvironment and immune response to favor survival and progression of the clone. Among IGH unmutated clones, BCR-transmitted signals provide predominantly proliferative signals, whereas IGH-mutated clones express gene pathways related to cellular anergy. The CLL cells continue to receive antigenic stimulation, and as the clone continues to proliferate, additional somatic mutations are acquired, and this may diminish the reliance of the clone on both antigenic stimulation and microenvironment support.
Some central questions to be investigated include the molecular basis of the 2:1 male:female gender bias in CLL, the mechanism by which some CLL acquire somatic mutations and others do not (ie, the molecular determinants of IGH mutation status), the mechanism by which preemergent CLL clones escape immune surveillance and deletion, and whether clonal evolution can be avoided through suppression of the proliferative compartment of CLL.
Finally, this scientific progress has created numerous targets of novel therapeutic interventions. Lineage-restricted cell surface molecules, such as ROR1 and CD37, are targets of novel MAbs. Lenalidomide has been shown to reverse the abnormal immunologic synapse formation in CLL, and also modulates the microenvironment through monocyte and NK cell activation. Novel small molecule inhibitors of BCR-mediated signaling are currently being tested in clinical trials. CAL101 selectively blocks a phosphatidylinositol 3-kinase isoform related to downstream signaling from CXCR4. Additional trials studying small molecular inhibitors of antiapoptotic proteins, such as BCL2, are underway. Elucidation of the mechanisms of chemotherapy resistance related to loss of p53 function has led to strategies to circumvent p53-dependent pathways. The advances in our fundamental understanding of the mechanisms of disease in CLL will undoubtedly lead to improved therapies for our patients.
Disclosures
Conflict-of-interest disclosure: The author has consulted and been affiliated with the Speakers Bureau for GlaxoSmithKline and Genentech, and has received research funding from Celgene and Eleos.
Off-label drug use: None disclosed.
Correspondence
Mark C. Lanasa, MD, PhD, Assistant Professor of Medicine, Division of Medical Oncology, Duke University Medical Center, DUMC Box 3872, 1 Trent Dr., Morris Building Room 25153, Durham, NC 27710; Phone: (919) 684-8964; Fax: (919) 684-5325; e-mail: mark.lanasa@duke.edu
