
Abstract
Chronic lymphocytic leukemia (CLL) is one of the most common lymphoid malignancies and is characterized by a tremendously variable clinical course. Additionally, whereas the median age at diagnosis is 72 years, CLL is diagnosed with increasing frequency in younger patients. Given the toxicities associated with currently available therapies, being able to predict which patients will need treatment could play a significant role in preserving bone marrow function and reducing morbidity and mortality. While a great many prognostic markers have been identified that predict outcomes for patients with CLL. Learning how to use these prognostic markers to provide patient care is more difficult.
Chronic lymphocytic leukemia (CLL) is one of the most common lymphoid malignancies in the United States, with an estimate of 15,490 new cases in 2009, resulting in 4,390 deaths.1 These data neglect to include the incidence of small lymphocytic lymphoma, which was formally recognized in 1994 as part of the Revised European-American Classification of Lymphoid Neoplasms to be the same disease as CLL,2 with an incidence of approximately 3950. Together, this translates into a prevalence of greater than 100,000 patients with CLL in the United States. With the vast majority of treatments carrying significant toxicities, knowing whom to treat and when becomes of paramount importance. As new chemoimmunotherapy regimens increase the potential for deep remissions, it is important to worry about the long-term toxicities of such treatments that might limit survival later in the course of the patient's disease.
The utility of prognostic factors can be assessed by their impact in three areas. First, prognostic markers need to not just reproducibly predict outcomes, but also note whether there is any benefit to being able to predict outcomes. In assessing prognostic markers, it is important to determine whether the information will aid in the care of the patient or just serve to increase anxiety about the patient's future.
Will the prognostic information change treatment? Will the information provided by prognostic information serve to induce anxiety without any benefit to the patient?
Patients with CLL are being diagnosed earlier in the course of their disease and living longer, leading to an increase in the variability in survival seen in CLL.3–4 Molica and Levato3 reported on patients diagnosed between 1970 to 1979, 1980 to 1989, and 1991 to 1998. They found an increase in the percentage of patients diagnosed with CLL in Binet Stage A at the time of their CLL diagnosis (26.3% vs 50.3% vs 72.0%, P < .0001), as well as an improvement in median overall survival (38 months vs 54 months vs 93 months; P < .0001) during the three time periods. But, when the survival data are stratified based on stage at diagnosis, the improvements in overall survival disappear for all but the Binet Stage C patients, with the patients diagnosed during 1970 to 1979 demonstrating a shorter survival compared with the other two time periods. Therefore, patients are living longer with their CLL diagnosis, resulting in longer time periods in which to determine when to start therapy. Additionally, contrary to prevailing ideas, almost 70% of patients die from causes related to CLL. This fact applies to older patients more than younger, as the relative survival for patients age 40 to 59 years is 69.9% versus 38.1% for patients greater than age 80 years.5 Therefore, there is great need to determine when to intervene, regardless of age.
The second aspect to explore is whether variables can be identified that can reliably and accurately predict prognosis. A quick review of the literature identifies over 35 different prognostic markers. Some of these are classified as “traditional,” whereas others are classified as “novel.” The traditional prognostic markers tend to be those obtainable from routine history, physical examination, and lab work. The novel prognostic factors tend to assess molecular aspects of the CLL cell themselves. (See Table 1 for a very partial list). With such a large number of possible factors, determining which ones are clinical useful is important.
A very partial list of prognostic markers
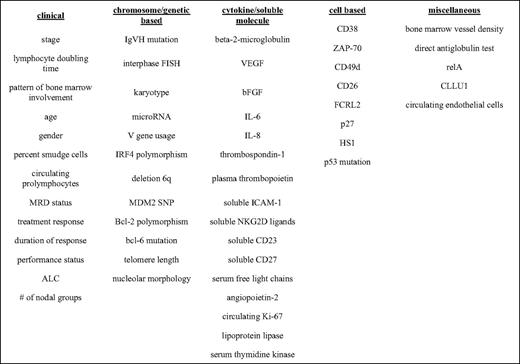
VEGF indicates vascular endothelial growth factor; bFGF, basic fibroblast growth factor; IL, interleukin; IRF4, interferon regulatory factor 4; MRD, minimal residual disease; SNP, single nucleotide polymorphism; ICAM-1, intercellular adhesion molecule 1; ZAP-70, zeta-associated protein-70.
The third aspect is whether the prognostic marker will impact on clinical decision making.
Traditional Prognostic Markers
The first prognostic marker to be delineated was the Rai stage.6 Published in 1975, the Rai Clinical Staging System utilized lymphadenopathy, organomegaly, and cytopenias (anemia and thrombocytopenia) to established five prognostic groups with median survivals of: (1) stage 0, > 150 months; (2) stage I, 101 months; (3) stage II, 71 months; (4) stage III, 19 months; and (5) stage IV, 19 months. It is important to note that the determination of lymphadenopathy and organomegaly was with physical examination only, not CT (computed tomography) scans. Whether the use of CT scans impacts on the survival seen for the individual Rai stages remains to be determined. An important feature of the Rai Clinical Staging System is that as patients progress to a higher stage, their prognosis parallels that of those who were diagnosed at that higher stage. These data have also been validated more recently by Molica et al.7 The Rai Clinical Staging System was later modified to include three groups, given the similarities between stages I and II and stages III and IV, with stage 0 being low risk, stages I and II being intermediate risk, and stages III and IV being high risk. The Rai model was followed by the Binet Staging System, which relied on the number of involved nodal areas involved by CLL and cytopenias to create a three-group classification.8 Both the Rai and Binet systems provide not only a prognosis for a patient, but also identify when a patient is appropriate for therapy. The National Cancer Institute-sponsored Working Group and the International Working Group on CLL guidelines both identify anemia and thrombocytopenia (Rai stage III/IV and Binet stage C) as indications for initiating therapy.
A great deal of variability remains with the individual stages. Other basic laboratory features that have been found to provide prognostic information include the following: pattern of bone marrow involvement,9,10 number of prolymphocytes in blood or bone marrow,11 age,12 gender,12 lymphocyte doubling time,13 absolute lymphocyte count,14 and β2-microglobulin.15 In an attempt to improve predictive power, Weirda et al16 developed a nomogram that merged age, β2-microglobulin, absolute lymphocyte count, sex, Rai stage, and number of involved lymph node groups into a single model. This model has subsequently been validated using the Mayo Clinic database, with estimated median survival times of not reached for low risk, 10.1 years for intermediate risk, and 7.2 years for high risk.17 For comparison, the median survival times by Rai stage for the patients used in Weirda's nomogram were: 11.5 years for stage 0, 11.0 years for stage I, 7.8 years for stage II, 5.3 years for stage III, and 7.0 years for stage IV.16 The differences in survival reported here compared with those from Rai's original paper represent the gains made over the intervening three decades.
Novel/Molecular-Based Prognostic Markers
It is not always clear whether a prognostic marker should fall into the “novel” or the “traditional” group, because some of the “traditional” factors were identified more recently than the “novel” factors. Regardless, the distinction between novel and traditional prognostic markers helps by designating those that are more likely to yield insights into the biology of CLL. The four novel prognostic markers that currently are in widespread use in clinical practice are the following: (1) immunoglobulin heavy-chain variable region (IgVH) mutational status, (2) interphase fluorescence in-situ hybridization (iFISH) abnormalities, (3) CD38, and (4) zeta-associated protein (ZAP)-70.
During germinal center reaction, the immunoglobulin genes of B cells undergo somatic hypermutation under the direction of T cells. This process helps to diversify the antibody repertoire, possibly improving an antibody's affinity for an antigen. Early studies indicated that CLL cells did not possess any somatic mutations in the complementarity-determining regions of its antibody genes. In 1992, work first suggested that cases of CLL may possess mutated or unmutated immunoglobulin genes.18 This idea was remarkable in that it suggested that CLL cases might be derived from two different stages of B-cell ontogeny, with the unmutated CLL cases being “preantigen” and the mutated CLL cases being “postantigen.” Work subsequently demonstrated correlations between trisomy 12 and deletion 13q14 interphase FISH abnormalities and IgVH mutation status. Once the germline immunoglobulin genes were fully characterized, it was possible to determine that approximately 40% of CLL cases were unmutated and 60% of cases mutated. In 1999, two groups published on the prognostic value of IgVH mutational status, with unmutated cases demonstrating a much more aggressive course and a median survival of 8 to 9 years versus > 20 years for those CLL cases with mutated IgVH genes.19,20
At the time, determination of the immunoglobulin gene mutational status was quite cumbersome. In attempt to identify more readily determined markers of CLL B-cell differentiation status, Chiorazzi and colleagues19 demonstrated that CD38 correlated with IgVH mutational status and predicted clinical outcome. Subsequent research confirmed the prognostic value of CD38 expression, but has questioned its ability to predict IgVH mutational status.21–23 Additional controversy has developed regarding whether CD38 expression can vary over the course of disease.24 Although it might be hypothesized that the changes in CD38 expression might be related to clonal evolution, data from deuterium-labeling experiments suggest that CD38 is transiently expressed on cells that are proliferating, with deuterium-labeled cells becoming CD38 negative and vice-versa. This conclusion is supported by seeing reciprocal changes in the percentages of deuterium-labeled cells during washout experiments and similar telomere lengths between the two fractions.25 Thus, CD38 represented an easily obtainable prognostic marker that correlated with IgVH mutational status and predicted prognosis independent of IgVH mutational status.
In the late 1990s, a novel technique for looking at chromosomal abnormalities was developed called iFISH. iFISH was well suited for use in CLL, given its low mitotic rate. In CLL, it is often difficult to obtain metaphase chromosomes and, when seen, they are possibly representing contaminating normal B cells in the sample. With iFISH, the number of chromosomal abnormalities seen in CLL increased from 51% to 82%.26,27 It is important to remember with iFISH that only the abnormalities being probed for are seen, whereas with karyotyping, the entire genome is examined. The original study by Dohner et al26 reported the presence of abnormalities in 82% of patients using probes designed to assess eight different regions. In the prognostic model outlined in the paper, patients were grouped into one of five groups, with median survivals of the following: (1) del 13q as a sole abnormality (133 months), (2) del 11q (79 months), (3) trisomy 12 (114 months), (4) del 17p (32 months), and (5) normal (111 months). All groups, except for del 13q, included samples that had multiple abnormalities. It is worth noting that “normal” does not indicate a normal karyotype, but rather the absence of one of the specified abnormalities tested for.
In 2001, attempting to identify genes that could distinguish mutated from unmutated cases of CLL, Rosenwald et al, 28 using gene array profiling, demonstrated that all CLL patients share a common CLL-specific gene expression signature, regardless of IgVH mutational status. Among the genes that were differentially expressed between mutated and unmutated CLL cases, ZAP-70 was the most differentially expressed gene between the two subtypes, being expressed in unmutated CLL. Several groups subsequently confirmed that ZAP-70 expression correlated with IgVH mutational status and predicted outcome for CLL patients.29–31 But, work done by Rassentiet al32 demonstrated that IgVH mutational status and ZAP-70 status were independent predictors of outcome, and, most significantly, in the discordant cases, ZAP-70 proved to be a stronger predictor of outcome compared with IgVH mutational status. The use of ZAP-70 as a tool in clinical practice to determine prognosis has been hampered by many technical issues, including methodology (flow cytometry vs immunohistochemistry), choice of monoclonal antibody used for detection, and means to permeabilize the cells given the internal location of the ZAP-70 protein.33,34 Further analyses suggest that the prognosis of discordant cases could be elucidated using other high-risk characteristics, such as V3–21 gene usage.35
Application of Prognostic Factors to Clinical Practice
The most important question left to answer is how the prognostic data discussed earlier can be used to guide clinical practice. The two decisions that need to be made prior to treating a patient are when and with which therapy. Regarding the timing of therapy, treatment is only indicated for those patients with documented active disease according to the International Working Group on CLL Treatment Guidelines.36 The only prognostic markers included in the criteria are Rai stage III or IV disease (progressive marrow failure) or a lymphocyte doubling time of less than 6 months.
Several studies have attempted to address whether initiating treatment at an earlier point in the disease course could have an impact on outcome.37–40 Although none of these trials demonstrated a benefit of early initiation of treatment compared with deferring treatment, these studies are hampered by two issues. First, these studies included a large number of patients with indolent disease who might not require treatment for long periods of time, if at all. These indolent cases were of sufficient number to make demonstrating an advantage of early intervention for high-risk populations difficult. Second, at the time of these trials, chlorambucil with or without prednisone was the only therapy available. With the more effective therapies now available, different results might be obtained.
Three studies have been initiated to try to revisit this question. The first trial, undertaken as an Intergroup trial involving the CALGB (Cancer and Leukemia Group B), ECOG (Eastern Cooperative Oncology Group), SWOG (Southwest Oncology Group), and NCIC (National Cancer Institute of Cancer) groups, stratified patients based on IgVH mutational status and was closed prematurely due to lack of accrual. The second study, the German CLL Study Group CLL1 trial, enrolled patients from 1997 to 2004 and stratified patients as high- or low-risk using thymidine kinase level > 7 U/L or β2-microglobulin > 3.5 mg/L and a lymphocyte doubling time < 12 months or diffuse bone marrow infiltration pattern. Preliminary data published at the 2007 American Society of Hematology meeting showed no difference in overall survival between the two groups after a median observation time of 45.3 months.14 The third study, sponsored by the German CLL Study Group, but conducted across Europe, is the CLL7 trial. The CLL7 trial randomizes Binet stage A patients with more than one adverse risk factor (unfavorable iFISH [del 17p, del 11q, trisomy 12], thymidine kinase level > 10 U/L, lymphocyte doubling time < 12 months, and unmutated IgVH gene) to FCR × six cycles versus observation. The choice of risk factors to identify the high-risk population for the CLL7 study was based on early results from the CLL1 study. The CLL7 trial completed accrual in January 2010.
The inability for the Intergroup trial to be completed is unfortunate. If one were to hypothesize a possible means for impacting on the poor prognosis of patients with high-risk disease, it might include reducing disease burden before CLL cells have time to acquire additional genetic changes that make them more resistant to therapy. An alternative hypothesis is that, because the secondary genetic changes are typically the result of therapy, avoiding treatment for as long as possible is advantageous.
The second decision involves the choice of therapy. Currently approved therapies for CLL include chlorambucil, fludarabine (or purine)-based chemotherapy, alemtuzumab, bendamustine, or ofatumumab. Extensive data demonstrate that 17p deleted or p53 mutated cases of CLL respond very poorly to purine analog or alkylator-based therapy.41–43 Alemtuzumab, on the other hand, appears to work via a p53 independent pathway, and has demonstrated efficacy in 17p deleted or p53 mutated CLL.44,45 But, whereas overall response rates of 64% in untreated or 40% are seen in fludarabine refractory CLL patients with deletion of 17p, median progression-free survival is short, ranging from 8 to 10.7 months. Similarly, in the fludarabine and alemtuzumab refractory CLL patients treated as part of the pivotal ofatumumab study, deletion of 17p did not predict for lack of response.46 These data do need to be taken cautiously, because p53 dysfunction could exist without deletion of 17p, and the numbers are small enough that they may lose statistical significance if grouped by p53 function. Although bendamustine does demonstrate some responses in fludarabine refractory CLL patients, the iFISH status was not reported as part of the trial.47 Therefore, in the setting of a CLL patient demonstrating p53 dysfunction, avoiding alkylator and purine analog-based therapies in favor of alemtuzumab, and arguably ofatumumab, would allow avoiding toxicities associated with therapies that have a low expectation of efficacy.
Another debate that exists regarding the treatment of CLL patients is whether FR (fludarabine plus rituximab) or FCR (fludarabine, cyclophosphamide, plus rituximab) should be the choice for initial therapy. Data from the German CLL Study Group CLL4 trial demonstrated improved response rates and progression-free survival, but no improvement in overall survival, for fludarabine (F) + cyclophosphamide (FC) as compared to F as initial therapy for patients with CLL.48 In subgroup analysis, deletion 11q was the only negative prognostic marker that remained predictive of improved progression free survival for FC, compared with F chemotherapy. But of even greater significance, overall survival was improved for the deletion 11q patients who received FC compared with those who received F chemotherapy. From these data, it could be concluded that, in patients with deletions of 11q, FC combination therapy must be utilized over F.
In my practice, I typically use FR as my firstline chemotherapy treatment for patients with CLL in the absence of deletion 11q or 17p. For those patients with deletion 11q, FCR is my preference based on data derived from the German CLL Study Group CLL4 trial. It is important to remember that there are no data available regarding FR versus FCR, and it is unknown whether rituximab might overcome some of the benefit of FC, compared with F. With regard to the patients with deletion 17p, if their disease is predominantly in the bone marrow, my preference is to utilize alemtuzumab. For those patients with deletion 17p and bulky lymphadenopathy, I favor FCR over alemtuzumab over concerns related to the lesser efficacy of alemtuzumab on bulky lymphadenopathy.
Conclusions
A great deal of work has led to the identification of a large number of prognostic markers for CLL patients. It is important to acknowledge one of the central tenets of statistics: “that statistics can be used to predict outcomes for a population, but not an individual.” Hence, when discussing prognostic information with patients, physicians can tell patients which survival curve they belong to, but not where on the curve they reside. Although prognostic information will provide many insights into the biology of CLL, the utility of this information in clinical practice is a separate question. We have begun to identify areas where these prognostic markers are useful in guiding choice of therapy, such as p53 dysfunction, and deletions of 17p and 11q. As more molecularly targeted therapies are developed, these biologically based prognostic markers are likely to become more influential. For now, we can only collect the information and wonder.
Disclosures
Conflict-of-interest disclosures: The author has received honoraria from Calistoga; research funding from and/or consulted for Calistoga, Genentech, GlaxoSmithKline, and Celgene; and been associated with the Speakers Bureau for Cephalon, Genentech, and GlaxoSmithKline.
Off-label drug use: None disclosed.
Correspondence
Richard R. Furman, MD, Assistant Professor of Medicine, Division of Hematology/Oncology, Weill Cornell Medical College, 525 East 68th St., New York, NY 10065; Phone: (646) 962-2064; Fax: (646) 962-1065; e-mail: rrfurman@med.cornell.edu
