
Abstract
Increased levels of fetal hemoglobin (HbF) can ameliorate the severity of the β-hemoglobin disorders, sickle cell disease (SCD) and β-thalassemia, which are major sources of morbidity and mortality worldwide. As a result, there has been a longstanding interest in developing therapeutic approaches for inducing HbF. For more than 3 decades, the majority of HbF inducers developed were based on empiric observations and have had limited success. Recently, human genetic approaches have provided insight into previously unappreciated regulators of the fetal-to-adult hemoglobin switch and HbF silencing, revealing molecular targets to induce HbF. This article reviews these developments and discusses how molecules including BCL11A, KLF1, MYB, SOX6, miRNAs 15a and 16-1, and histone deacetylase 1 and 2 (HDAC1/2) could be important targets for HbF induction in humans. The current understanding of how these molecules function and the benefits and drawbacks of each of these potential therapeutic targets are also examined. The identification of these regulators of HbF expression is extremely promising and suggests that rationally designed approaches targeting the very mechanisms mediating this switching process could lead to better, less toxic, and more effective strategies for HbF induction.
Introduction
The β-hemoglobin disorders, sickle cell disease (SCD) and β-thalassemia, are major causes of morbidity and mortality worldwide,1 as the most common genetic disorders in the world. With the epidemiological transition that many developing countries are undergoing, resulting in significant decreases in mortality from infectious and nutritional causes, there will likely be an increase in the prevalence of these diseases in the ensuing years. In SCD, a single base substitution causes a missense mutation of a valine for glutamic acid at amino acid 6 of the β-globin protein chain, which leads to a propensity of the sickle hemoglobin to polymerize. This results in deformation of RBCs containing this hemoglobin, which can block small blood vessels, leading to impaired oxygen delivery to tissues. This can cause significant pain crises, respiratory complications, and organ damage. In β-thalassemia, insufficient production of the β-globin molecule results in an excess of unpaired α-globin chains that can precipitate within erythroid precursors. The precipitation of these free α-globin chains impairs the maturation of these precursors, leading to their death and thereby causing ineffective production of RBCs. Significant anemia results and the consequent expansion of erythroid precursors can lead to secondary problems in bones and other organs.
Whereas the only cures for these disorders involve BM transplantation2 and gene or cellular therapy,3 there are numerous challenges associated with these treatments and therefore they have significant limitations for widespread use, particularly in the developing world.4 These curative therapies still remain largely experimental, although important advances in these approaches are being made.5 Currently, the major treatment for SCD and β-thalassemia involves symptomatic care and transfusion of RBCs as clinically necessary. However, the use of regular transfusion therapy can lead to significant problems, particularly iron overload, which can even occur when the strictest iron-chelation regimens are used.6
Treating the β-hemoglobinopathies through HbF induction
A series of important natural observations demonstrated that the severity of SCD and β-thalassemia could be ameliorated via increased production of fetal hemoglobin (HbF), which is composed of the β-like globin chain, γ-globin, and the adult α-globin molecule. In normal infants, the predominant β-like globin before birth is γ-globin, and there is a switch to adult β-globin expression in the months after birth, a process known as the fetal-to-adult hemoglobin switch (Figure 1).7,8 Children with SCD were noted to be asymptomatic until after infancy, which was postulated to be attributable to elevated HbF levels.9 This notion was substantiated by observations of patients with compound heterozygosity for SCD and hereditary persistence of HbF mutations, who were largely asymptomatic.10 Moreover, certain populations of patients with SCD, such as those from parts of Saudi Arabia and India, had unusually high levels of HbF associated with a less symptomatic clinical course.10–12 These observations were confirmed with larger epidemiological studies showing that increased HbF levels can significantly ameliorate the clinical severity of SCD.13–15 Similar observations were made in patients with β-thalassemia. Clinical observations in rare β-thalassemia patients with highly elevated levels of HbF showed that these increased levels resulted in a milder clinical course, and infants with β-thalassemia only begin to show symptoms after the expression of HbF declines in the months after birth.10,16 Larger epidemiological studies of thalassemia populations have confirmed these findings.17–19

Molecular regulation of the fetal-to-adult hemoglobin switch. The human β-globin gene locus, which is located on chromosome 11, has a series of 5 genes that are developmentally expressed at distinct stages of human ontogeny (panel A bottom). The ε-globin gene is restricted to the transiently expressed embryonic primitive lineage of RBC produced in the first several weeks of gestation. The γ-globin genes encode the genes that uniquely form HbF, which is the predominant hemoglobin during the course of gestation. There is a switch to expression of the adult β-globin gene around the time of birth (B). B-cell lymphoma/leukemia 11A (BCL11A) and its protein partners, including GATA binding protein 1 (GATA1), zinc finger protein multi-type 1 (ZFPM1 or FOG1), and the nucleosome remodeling and deacetylase (NuRD) complex, bind to sequences within the globin locus (panel A tan ovals) and repress the expression of the γ-globin genes. Krüppel-like factor 1 (KLF1) interacts with this process by positively regulating the expression of BCL11A (green arrow in panel A) and also by directly binding to and promoting transcription of the adult β-globin gene. It is unknown whether KLF1 affects HbF expression through more global effects on erythropoiesis (panel A top), which may also be targeted by other factors associated with HbF (eg, MYB). HSs indicates DNase I–hypersensitive sites; LCR, locus control region. (Reprinted with permission from Sankaran and Nathan.7 )
Molecular regulation of the fetal-to-adult hemoglobin switch. The human β-globin gene locus, which is located on chromosome 11, has a series of 5 genes that are developmentally expressed at distinct stages of human ontogeny (panel A bottom). The ε-globin gene is restricted to the transiently expressed embryonic primitive lineage of RBC produced in the first several weeks of gestation. The γ-globin genes encode the genes that uniquely form HbF, which is the predominant hemoglobin during the course of gestation. There is a switch to expression of the adult β-globin gene around the time of birth (B). B-cell lymphoma/leukemia 11A (BCL11A) and its protein partners, including GATA binding protein 1 (GATA1), zinc finger protein multi-type 1 (ZFPM1 or FOG1), and the nucleosome remodeling and deacetylase (NuRD) complex, bind to sequences within the globin locus (panel A tan ovals) and repress the expression of the γ-globin genes. Krüppel-like factor 1 (KLF1) interacts with this process by positively regulating the expression of BCL11A (green arrow in panel A) and also by directly binding to and promoting transcription of the adult β-globin gene. It is unknown whether KLF1 affects HbF expression through more global effects on erythropoiesis (panel A top), which may also be targeted by other factors associated with HbF (eg, MYB). HSs indicates DNase I–hypersensitive sites; LCR, locus control region. (Reprinted with permission from Sankaran and Nathan.7 )
The human globin genes were among the first genes to be cloned, and soon thereafter the structure of the entire β-globin cluster was mapped and sequenced. Around this time, it was observed that the silenced γ-globin genes in the β-globin cluster underwent DNA methylation in adult erythroid cells, whereas this was not the case in erythroid cells from embryonic life.4 Given these observations, preclinical trials in primates of the DNA-hypomethylating agent 5-azacytidine were subsequently performed and demonstrated success at inducing HbF.4,20 This approach was then tested in patients with β-thalassemia and SCD.21 However, there was concern over the long-term consequences of a potentially mutagenic compound such as 5-azacytidine. Around this time, it was suggested that the effect of 5-azacytidine on HbF induction might relate, at least in part, to its ability to act as an S-phase cell-cycle inhibitor, rather than through alteration of DNA methylation at the γ-globin gene promoters.4 Studies in primates demonstrated that numerous S-phase inhibitors, including hydroxyurea, were all highly effective as inducers of HbF. This then led to small clinical trials of hydroxyurea, which had the safest side-effect profile, in SCD patients in whom a robust HbF inductive response was demonstrated.22 The use of hydroxyurea was then examined in larger clinical trials that led to its US Food and Drug Administration approval for use in adult patients with SCD.23,24 Unfortunately, hydroxyurea is only effective to some extent in a subset of patients with SCD. There has also been limited success with the use of hydroxyurea in β-thalassemia, which may be attributable to the requirement for much more HbF to achieve globin chain balance in this disease and to the inability to escalate hydroxyurea dosing as a result of cytopenias.
In the mid-1980s, it was shown that infants of diabetic mothers have a delayed fetal-to-adult hemoglobin switch.25,26 Because it was known that hydroxybutyrate is elevated in these diabetic mothers, a series of experiments was performed to determine whether butyrate or other similar short-chain fatty acids may be effective as inducers of HbF. Whereas initial trials showed promise,27,28 these therapies were not widely effective in larger clinical trials.4,29 It should be noted nonetheless that a subset of patients did show responses in these trials.4,29 However, additional concerns were raised over possible toxic effects and the difficulty in delivery of these medications, which limited further clinical trials and drug-development efforts. It has been suggested that these short-chain fatty acids are likely to induce HbF through the inhibition of histone deacetylases (HDACs).30 A variety of HDAC inhibitors can be potent inducers of HbF in vitro, and so it is possible that these compounds may show clinical efficacy in vivo.31,32
Identifying molecular targets for HbF induction
Molecules that regulate the fetal-to-adult hemoglobin switch and help to maintain HbF silencing in humans have been long sought given the possibility of using this information to develop targeted therapeutic approaches for HbF induction.7,32 Whereas numerous regulators of erythropoiesis and globin gene regulation had been identified, none of the previously examined molecules appeared to be specific regulators of the fetal-to-adult switch. Recent insight into this process has come from studies using human genetic approaches. By taking advantage of natural variation in HbF levels in humans, genome-wide association (GWA) studies were performed to find common genetic variants associated with variations in HbF levels. The initial GWA studies of HbF were performed in nonanemic individuals. Two separate studies were performed around the same time and showed association of variants at 3 major genomic loci with HbF levels.33,34 This included the β-globin locus on chromosome 11, a region between the genes HBS1L and MYB on chromosome 6, and a region within the BCL11A gene on chromosome 2. These findings were replicated in patients with SCD and β-thalassemia, and the ameliorating effect of these loci upon these diseases was also demonstrated in multiple other studies.18,19,33,35
Regulation of HbF expression by BCL11A and SOX6
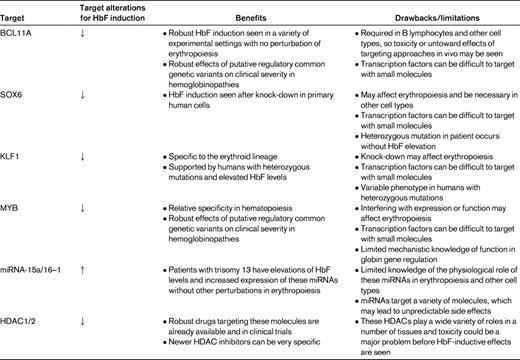
The role of BCL11A was described previously for B-lymphocyte production, but no studies had explored whether this gene had a role in erythropoiesis. As a result, the role of this gene in human globin gene regulation and erythropoiesis was assessed soon after the initial association studies.36 BCL11A protein levels appeared to be correlated with the developmental stage of expression, such that primitive and fetal liver erythroid cells that expressed high levels of γ-globin had low or absent expression of the full-length forms of BCL11A, whereas shorter variant forms of the protein were noted in these cells. This result suggested that this gene product may act as a repressor of the γ-globin genes. To directly test this, knock-down of BCL11A using siRNA approaches was performed in primary adult erythroid progenitors, and γ-globin expression could be induced. The degree of γ-globin induction appeared to be linearly related to the extent of knock-down of BCL11A. Interestingly, major perturbations in erythropoiesis did not appear to occur despite the robust γ-globin induction seen. Follow-up studies have demonstrated similar results when knocking down the expression of BCL11A using siRNAs.37–39 It was also shown that BCL11A directly interacts with chromatin at the human β-globin locus in primary erythroid cells and that it appeared to act as part of a complex with the transcription factor GATA-1 and the NuRD chromatin remodeling and repressor complex (Figure 1).36 The NuRD complex includes HDAC1 and HDAC2, which have been suggested to be the critical HDACs necessary for HbF silencing (Table 1).40
Whereas hemoglobin switching in mouse models, even those harboring a human β-globin locus transgene, appears to diverge from the normal ontogeny of globin expression seen in humans, an evolutionarily conserved role of BCL11A in globin gene silencing and switching was demonstrated in mice.41 Specifically, transgenic mice with the human β-globin locus express the γ-globin gene in a manner similar to the endogenous mouse embryonic globin genes, although there is some variation in the expression between these globin genes.42 Mice lacking BCL11A appear to have normal erythropoiesis, but fail to silence the embryonic globin genes in definitive erythroid cells, allowing persistent expression of γ-globin when the intact human β-globin locus is present.41 This study reinforces the importance of BCL11A as a critical mediator of globin switching in mammals.
The exact mechanism by which BCL11A silences γ-globin expression remains unclear. A recent study suggests that this may be mediated through both interactions with transcription factors such as SOX6 that bind chromatin at the proximal γ-globin promoters and through long-range interactions with a variety of regions throughout the β-globin gene cluster.43 When BCL11A is absent, the conformation of the β-globin locus changes such that the upstream enhancer known as the locus control region is juxtaposed with the transcriptionally activated γ-globin genes. A similar phenomenon occurs when cells are treated with HDAC inhibitors that induce γ-globin gene expression.40 However, it is unclear whether these conformational alterations are directly mediated by BCL11A or if they occur secondary to the HbF-inductive effect of BCL11A (or HDAC) inhibition. Nonetheless, these findings strongly support the notion that BCL11A has a direct role in silencing γ-globin expression within the β-globin locus. Therefore, BCL11A is likely to be an important therapeutic target. The fact that it can induce HbF without resulting in a major perturbation in erythropoiesis is very promising, although it is known to have important effects in other lineages such as B lymphocytes, stressing the importance of in vivo modeling and analysis as part of ongoing efforts to target BCL11A for HbF induction (Table 1). Strategies aimed at targeting BCL11A may rely on altering its expression level or activity. SOX6 may also be a potentially promising target for HbF induction,43 although it is known to be necessary for normal erythropoiesis (Table 1).44 Interestingly, however, a patient has been reported with a heterozygous disruption of SOX6 who did not have elevated HbF levels, suggesting that the SOX6 gene may have a mechanism for dosage compensation or that there may be a particular threshold necessary for reducing the expression of this gene to have robust HbF induction.45
Silencing of HbF by KLF1
Using independent and complementary approaches, two studies have demonstrated that the expression of BCL11A appears to be controlled by the erythroid-specific transcription factor KLF1. In one study, a mouse with a hypomorphic allele of KLF1 had elevations of embryonic globin expression, and transgenic mice with the human β-globin locus showed persistent expression of γ-globin.39 As a result, the investigators used siRNA approach to find out whether the same regulation could occur in primary human erythroid cells and demonstrate a similar connection. They were able to demonstrate that this effect occurs through both direct effects of KLF1 at the β-globin locus, but also via indirect effects mediated through reduced expression of BCL11A upon KLF1 knock-down. This finding demonstrated that KLF1 was a direct positive transcriptional regulator of BCL11A expression. A different study examined the genetic basis of an unlinked form of hereditary persistence of HbF that was found to be due to a KLF1 missense mutation in this family.38 Using primary cells from these patients and from unaffected controls, the investigators of that study were able to demonstrate that the effect seen was in part attributable to a silencing effect of KLF1 on BCL11A.
One area of ongoing uncertainty relates to the human phenotypes seen in various cases of KLF1 mutations. Whereas in the initial report, all recipients of the missense mutation had elevations in HbF expression, it should be noted that this actually varied between 3% and 19% of total hemoglobin levels.7 In addition, other reports of heterozygous KLF1 mutations in humans either show concomitant disruption of erythropoiesis or little effect on HbF expression.46,47 The basis of this variation remains to be determined and will be important to better understand the mechanisms by which KLF1 acts, both directly and indirectly, to affect HbF expression (Figure 1). This will be critical for any future work attempting to target KLF1 for HbF induction, particularly if the untoward effects of such inhibition on erythroid differentiation are to be avoided. However, given the specificity of KLF1 within the erythroid lineage, and if disruption of erythropoiesis could be avoided while allowing for HbF expression, it may be a very promising therapeutic target to induce HbF (Table 1).
The role of MYB and miRNAs 15a/16-1 in HbF regulation
The GWA studies also demonstrated strong associations of variants in the intergenic region between the genes HBS1L and MYB with HbF levels. The exact mechanism by which these variants result in elevated HbF levels has remained unclear, although it has been suggested that this may occur through an effect on the expression or function of MYB.7,48 Overexpression of MYB in K562 cells reduced γ-globin expression, and primary erythroid cultures with higher HbF expression had lower levels of MYB expression.49 More direct evidence lending further support to a connection between MYB and HbF expression has recently been reported. Following up on a classic clinical observation that patients with a trisomy of chromosome 13 have elevations of HbF, and using partial trisomy cases to map this trait, it was shown that a gene in 13q14 appears to be responsible for mediating this effect.50 Using an integrative genomic approach, 2 co-transcribed miRNAs, 15a and 16-1, appeared to be top candidates as mediators of this trait. This was supported by the finding that slight overexpression of these miRNAs in primary adult erythroid cells could result in elevations of γ-globin expression. Consistent with prior studies,51 these miRNAs appeared to potently target and down-regulate MYB protein expression. Finally, siRNA-mediated knock-down of MYB could result in a similar effect, suggesting a direct connection between the effect of these miRNAs on HbF silencing and their effect on MYB expression. This finding also supports the idea that the intergenic variants between HBS1L and MYB may act to effect MYB expression.48 Sequencing of MYB has revealed rare coding variants within this gene that appear to be associated with elevated HbF levels in patients with SCD, providing further support for a role of MYB in modulating HbF expression.52
Numerous studies have shown that MYB is necessary for normal erythropoiesis, although hypomorphic mouse alleles of this gene have subtle effects on erythropoiesis.53 Moreover, such in vivo studies demonstrate the specificity of MYB in hematopoietic cell types. If the function of MYB could be partially disrupted and targeted in an erythroid-specific manner, then it may be an attractive therapeutic target (Table 1). The exact physiological role of miRNA-15a and miRNA-16-1 in erythropoiesis remains to be elucidated; however, it is possible that these miRNAs may be attractive targets for HbF induction. Support comes from the finding that trisomy 13 patients have relatively normal erythropoiesis while having persistent HbF expression.50 Much more work is necessary to elucidate the mechanisms through which these factors affect HbF expression, which will be important in strategies for HbF induction.
Other factors involved in HbF regulation
In addition to the aforementioned molecules, a few other factors have been suggested to play a role in HbF expression, although support from human genetic studies is not currently present for these other factors. One recent study described the role of the Friend of PRMT1 (FOP1) protein in HbF silencing.54 Knock-down of this factor in primary human adult erythroid cells resulted in robust elevations of HbF levels. The mechanism through which this factor carries out this activity remains unclear and further work will be necessary to more carefully investigate this observation. In addition, other factors such as the orphan nuclear hormone receptors TR2, TR4, and COUP-TFII have been suggested to play a role in HbF silencing.8 Further work to examine the role of these factors in HbF regulation will be needed to better understand how these molecules regulate this process.
As mentioned above, the HDACs are promising targets and are recruited by and interact with a variety of transcription factors involved in globin gene regulation, such as BCL11A.8 Other DNA- and histone-modifying enzymes have been suggested to play a role in globin gene regulation and thus are worthwhile considerations as possible targets for HbF induction. For example, there is support for a role of the DNA methyltransferase DNMT1 in silencing HbF.55 Because 5-azacytidine and its derivative, decitabine, which both induce HbF in vivo, target DNMT1, it is possible that future strategies targeting this pathway may lead to more effective therapies.
Approaches and future directions
Whereas the recent findings described above have uncovered promising molecular targets for therapeutic efforts aimed at HbF induction, a great deal of further work is necessary before these findings can be translated to therapeutic advances. The question of how best to develop such therapies is of great interest in this field. One immediately applicable approach would be to use gene therapeutic approaches to deliver siRNAs that target the molecules described above. With the exception of miRNAs 15a and 16-1, the other therapeutic targets discussed here all appear to have HbF-inductive effects when their expression is reduced (Table 1). Preliminary preclinical findings support such approaches.37 In addition to or in place of delivery of a nonmutated globin gene, such siRNA molecules could be extremely useful. These gene therapy methods are promising and could be targeted to the hematopoietic system alone. Given recent advances in this field,56 it is possible that such approaches could enter the clinical arena in the near future. Such gene therapy trials that are performed on a relatively small scale may help to reveal which therapeutic targets would be efficacious in vivo for more broadly applicable small-molecule approaches. Another approach would be to introduce exogenous molecules such as zinc-finger transcription factors, which interfere with the normal activity of endogenous molecules found at the β-globin locus and thus act to induce HbF.37
Small molecules would be ideal as therapies in these globally widespread diseases. However, nearly all of the molecular targets that currently exist are transcriptional regulators of globin gene expression (Table 1). These molecules have been notoriously difficult to target with small molecules, although recent approaches suggest that promising leads could be developed.57 As enzymes, the HDACs are ideal targets, and many specific inhibitors of HDACs have already been developed. Some of these appear to be promising leads for HbF-inductive therapies and are already in clinical trials for other indications such as cancer.58 Other small molecules targeting epigenetic enzymes, such as DNMT1, may show possible benefit as HbF inducers and, in many cases, inhibitors of these enzymes are already available or are in development. At the present time, it is difficult to predict which targets may be best, and, given the number of recently identified molecular targets, it is likely that the broadest approaches aimed at targeting as many of these molecules as possible may eventually yield specific candidates that can successfully pass the early stages of evaluation. However, the fact that there are so many recently identified targets lends promise to this field. In addition, the ongoing basic science studies that are aimed at better understanding the mechanisms by which such molecules silence HbF will likely yield additional insights into how this process can best be modulated.
Acknowledgments
I apologize to those investigators whose work could not be cited here because of space limitations. I am grateful to my mentors who have given me the opportunity to study hemoglobin switching and to the many colleagues I have had the privilege of working with.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Vijay G. Sankaran, Children's Hospital Boston, 300 Longwood Avenue, Boston, MA 02115; Phone: (617) 355-6000; e-mail: sankaran@broadinstitute.org.
