
Abstract
The ability of immune-competent donor T cells to mediate a beneficial graft-versus-leukemia (GVL) effect was first identified in the setting of allogeneic hematopoietic stem cell transplantation (allo-HSCT) for hematologic malignancies. Unfortunately, with the exception of chronic myelogenous leukemia and EBV-induced lymphoproliferative disease, allo-HSCT GVL lacks the potency to significantly affect disease progression or recurrence in most other hematologic malignancies. The inadequacy of a GVL effect using past approaches is particularly evident in patients with lymphoid malignancies. However, with the advent of improved gene transfer technology, genetically modified tumor-specific immune effectors have extended cellular immunotherapy to lymphoid malignancies. One promising strategy entails the introduction of genes encoding artificial receptors called chimeric antigen receptors (CARs), which redirect the specificity and function of immune effectors. CAR-modified T cells targeted to the B cell–specific CD19 antigen have demonstrated promising results in multiple early clinical trials, supporting further investigation in patients with B-cell cancers. However, disparities in clinical trial design and CAR structure have complicated the discovery of the optimal application of this technology. Recent preclinical studies support additional genetic modifications of CAR-modified T cells to achieve optimal clinical efficacy using this novel adoptive cellular therapy.
Introduction
Leukemia is the most common pediatric malignancy, accounting for 31% of all pediatric cancers diagnosed in the United States for children less than 15 years of age and 25% in patients less than 20 years of age.1 Acute lymphoblastic leukemia (ALL) accounts for the majority of cases (23% of all pediatric cancer) and is predominantly of B-cell origin (approximately 85% of ALL cases). In 2012, it is estimated that more than 6000 cases of ALL will be diagnosed in adults and children, with approximately two-thirds occurring in children.2 Acute myelogenous leukemia (AML) occurs less commonly in children, at 4% of all pediatric cancers diagnosed, but still represents approximately 20% of new cases of pediatric leukemia.1 The majority of the estimated 14 000 cases of AML diagnosed each year in the United States will be in adults.2
Most pediatric patients with B-cell acute lymphoblastic leukemia (B-ALL) are cured with standard chemotherapy regimens, with overall survival exceeding 80% in many reported series.3–6 For some patients with very-high-risk features, the use of allogeneic hematopoietic stem cell transplantation (allo-HSCT) in first complete remission is recommended.7 In contrast, most adult patients diagnosed with ALL have a poor prognosis, with survival rates < 40% in most series.8 For pediatric patients with AML, the chance of cure with chemotherapy alone is significantly less, with only 50%-65% of patients achieving long-term survival.9–12 Despite optimal therapy, long-term survival is less likely in adults, with older patients having a worse prognosis.13,14 Recommendations have been made for the use of allo-HSCT as a consolidative treatment for both adult and pediatric patients with high-risk features or with a suitable matched related donor.15,16 Unfortunately, the survival of both adult and pediatric patients with relapsed or refractory ALL or AML remains dismal.17–23 In fact, even with advances in chemotherapy or the use of allo-HSCT, the leading cause of mortality for children diagnosed with cancer is relapsed leukemia.1 To address the problem of limited therapeutic success with current options for treatment of refractory or relapsed leukemia, novel adoptive cellular therapies have been developed.
allo-HSCT
To date, allo-HSCT is the most common and successful example of cellular therapy for leukemia. Conditioning chemotherapy and/or radiation provides sufficient immunosuppression of the recipient to prevent donor HSC rejection and facilitates the destruction of residual malignant cells. Subsequent infusion of HSCs from healthy donors provides a hematologic and immunologic recovery for recipients. Optimizing the balance between antileukemic efficacy and patient toxicity has been a long-term objective of clinical BM transplantation research. Significantly, the antileukemic effect of allo-HSCT is not limited to the cytoreductive regimen. Immunocompetent donor T cells can mediate a beneficial GVL effect facilitated through the recognition of allo-Ags presented on residual tumor cells by donor T cells. Evidence to support the GVL of allo-HSCT was first demonstrated by Weiden et al in patients with acute leukemia.24 In that landmark paper illustrating the GVL effect of allo-HSCT, higher relapse rates were seen in patients with syngeneic donors compared with patients with allogeneic donors who experienced acute and/or chronic GVHD.24 Evidence supporting the presence of a GVL of allo-HSCT includes higher relapse rates in patients with chronic myelogenous leukemia after T cell–depleted allo-HSCT and anecdotal reports of remissions achieved in patients with relapsed/refractory leukemia after allo-HSCT following withdrawal of immune suppression or a GVHD flare.25–28 Unfortunately, attempts to enhance this GVL benefit (eg, using HLA disparate donors) has been met with the untoward consequences of increasing GVHD with its associated morbidity and mortality, thus counteracting the associated potential GVL benefit derived from allo-HSCT.
Recommendations for the use of allo-HSCT in both adult and pediatric patients with ALL and AML have been described previously.7,15,16,29 HLA matched related donors are the favored source of HSCs for allo-HSCT. Unfortunately, the majority of patients requiring allo-HSCT do not have an appropriately HLA matched relative. Alternative HSC sources include: unrelated donors, umbilical cord blood (UCB), or haploidentical related donors. The use of alternative HSC sources for allo-BMT has expanded the pool of eligible patients markedly. Recent reports have shown equivalent leukemia-free survival after UCB transplantations compared with BM transplantations in children and UCB transplantations vs peripheral blood stem cell or BM transplantations in adults.30,31 Haploidentical related donor transplantation has gained renewed interest after reports of improved engraftment and reduced GVHD after posttransplantation cyclophosphamide (in the setting of reduced intensity conditioning) or improved methods of T-cell depletion through CD34+ selection (in the setting of myeloablative conditioning).32–34
Donor leukocyte infusion
One approach that attempts to use the GVL effect of donor T cells is donor leukocyte infusion (DLI) after allo-HSCT. In the setting of relapsed chronic myelogenous leukemia after allo-HSCT, additional therapy with DLI has demonstrated the most compelling direct evidence of a clinically meaningful GVL effect.35–37 In addition, in the setting of EBV-induced lymphoproliferative disease (EBV-LPD), DLI from EBV-seropositive donors or the infusion of EBV-specific cytotoxic T cells (EBV-CTLs) generated ex vivo can induce durable disease remissions by restoring cell-mediated viral immunity.38–40 However, in the setting of relapsed adult and pediatric acute leukemia after allo-HSCT, the use of DLI has only rarely induced durable remissions, is associated with a high incidence of acute and chronic GVHD, and does not provide a meaningful survival benefit for the majority of patients.36,37,41,42
NK-cell infusion
The identification of a GVL effect mediated by alloreactive natural killer (NK) cells was first described in the studies by Ruggeri et al.43 This phenomenon is most evident in the setting of haploidentical transplantations for AML in which donor NK cells improve transplantation outcomes by reducing leukemia relapse in the setting of killer cell Ig–like receptor ligand mismatches, thus triggering donor NK cell versus recipient leukemia alloreactivity.43 Several centers have attempted to use this apparent GVL effect of alloreactive NK cells in clinical trials through the infusion of haploidentical NK cells, primarily in patients with either poor-risk or relapsed AML.44,45 To date, reported beneficial clinical outcomes have been reported in a minority of treated patients.44,45
CARs: genetically engineered adoptive cell therapy
Conventional approaches to adoptive cellular therapy of leukemia, including allo-HSCT and DLI, in most cases mediate at best a modest GVL effect. Further, this modest antitumor effect is offset by the significant risk of donor T cell–mediated GVHD, which has diminished enthusiasm for these cellular therapeutic approaches. To address the problem of limited success with associated significant toxicity, novel cellular therapies using genetically modified, tumor-specific T cells have gained significant interest among investigators.
With the advent of enhanced gene-transfer technology, especially in the arena of gamma-retroviral and lentiviral vector design, it is now possible to efficiently introduce tumor-specific cloned TCR genes46,47 or genes encoding chimeric Ag receptors (CARs)48 into immune effector cells. Through this genetic reprogramming, these immune effector cells are redirected to target Ags expressed by leukemic cells. In most clinical applications to date, a patient's own T cells may be reprogrammed to express these tumor-specific receptors, largely minimizing the potential risk of adverse GVHD-related toxicities as seen in the setting of allo-HSCT and DLI.
The use of tumor-targeted TCR gene transfer has to date had limited clinical application in the treatment of hematological malignancies. This approach is further limited by complications including: a lack of efficient cloned TCR expression49 ; theoretical mismatched pairing of cloned alpha- and beta-chains with endogenous alpha- and beta-chains, resulting in highly unpredictable and potentially autoimmune TCR pairings50 ; and the HLA-restricted nature of TCR recognition that limits the application of this technology to specific HLA repertoires.
In contrast, genetic modification of immune effectors via a CAR is not restrained by these limitations and has several advantages. Because tumor targeting is HLA independent, the use of CARs is applicable to a broad range of patients irrespective of HLA phenotype. Furthermore, CAR modification of immune effectors overcomes the tumor's ability to escape immunodetection by down-regulation of HLA molecules on the cell surface. Targeting of tumor Ags by CAR-modified T cells is applicable to any cell-surface Ag, including proteins, carbohydrates, and glycolipids, for which a mAb can be generated.51 This enables CAR-modified T cells to respond to a broader range of targets compared with TCR-modified immune effector cells. Finally, the ability to generate a large quantity of tumor-specific T cells in a relatively short period of time makes this strategy feasible for use in the clinical setting.52
CARs are composed of an Ag-specific binding domain (most commonly a single-chain–variable fragment derived from the fused variable heavy- and light-chain domains of a tumor-targeted mAb) fused to a transmembrane domain followed by one or more cytoplasmic signaling domains (Figure 1). In initial designs, CARs were designed to contain a single cytoplasmic signaling domain derived most commonly from the TCR-derived CD3ζ chain. These “first-generation” CARs, when expressed in T cells, mediate a primary activation signal upon encounter with the targeted Ag (termed signal 1), but in the absence of additional costimulation (ie, signal 2) undergo activation-induced apoptosis or senescence, termed anergy.53 To address this limitation, we and others further modified CAR structure to add additional cytoplasmic signaling domains, including T cell–costimulatory signaling domains (eg, CD28, 4-1BB, or OX-40), resulting in “second-generation” CARs. These second-generation CARs containing costimulatory signaling domains are capable of delivering both signal 1 and signal 2 upon encounter with the targeted tumor Ag.54–58 Given the promising preclinical results with T cells modified to express second-generation CARs, investigators have favored this CAR design in initial clinical trials. Further modification of CAR design includes adding more than one costimulatory signaling domain in addition to the CD3ζ chain, resulting in “third-generation” CARs; this hasalso been translated to the clinical setting (Figure 1).59–62

CAR technology evolution through the generation of more potent CARs. First-generation CARs classically contain only one signaling domain, typically the cytoplasmic signaling domain of the CD3 TCRζ chain. Second-generation CARs containing 2 signaling domains typically include the addition of the cytoplasmic signaling domains of the costimulatory receptors CD28, 4-1BB, or OX-40, among others. Third-generation CARs attempt to harness the signaling potential of 2 costimulatory domains: classically, the CD28 domain followed by either the 4-1BB or OX-40 signaling domains. CAR-modified T-cell potency may be further enhanced through the introduction of additional genes, including those encoding proproliferative cytokines (ie, IL-12) or costimulatory ligands (ie, 4-1BBL), thus producing “armored” fourth-generation CAR-modified T cells.
CAR technology evolution through the generation of more potent CARs. First-generation CARs classically contain only one signaling domain, typically the cytoplasmic signaling domain of the CD3 TCRζ chain. Second-generation CARs containing 2 signaling domains typically include the addition of the cytoplasmic signaling domains of the costimulatory receptors CD28, 4-1BB, or OX-40, among others. Third-generation CARs attempt to harness the signaling potential of 2 costimulatory domains: classically, the CD28 domain followed by either the 4-1BB or OX-40 signaling domains. CAR-modified T-cell potency may be further enhanced through the introduction of additional genes, including those encoding proproliferative cytokines (ie, IL-12) or costimulatory ligands (ie, 4-1BBL), thus producing “armored” fourth-generation CAR-modified T cells.
Adoptive therapy with CD19-targeted, CAR-modified T cells: rationale and preclinical studies
Genetically engineered tumor-targeted cell therapy first and foremost requires the identification of a suitable target Ag to avoid the toxicity associated with targeting Ags expressed on normal tissues. To this end, most clinical trials in the field of cellular therapy for leukemia have targeted the CD19 Ag. Although CD19 is expressed on normal B cells and most B-cell malignancies, including B-ALL, chronic lymphocytic leukemias (CLL), and most non-Hodgkin lymphomas, the Ag is not expressed on normal HSCs. Whereas optimal immune targeting of the CD19 Ag will result in the eradication of B-cell tumors and may induce subsequent B-cell aplasia, successful targeting of this Ag will otherwise spare other normal hematopoietic precursors and BM function.
Early preclinical studies using adoptive transfer of CD19-targeted, CAR-modified human T cells into CD19+ human tumor–bearing immunocompromised mice demonstrated marked antitumor responses mediated by these genetically modified T cells.58,63 In addition, preclinical studies demonstrated superior proliferative and antitumor activity, both in vitro and in vivo, of T cells modified to express second-generation CD19-targeted CARs containing either the costimulatory CD28 or 4-1BB cytoplasmic signaling domains.58,64 Given these promising preclinical findings, several centers, including our own, have translated this technology to the clinical setting as phase 1 clinical trials treating patients with CD19+ malignancies (Table 1).
Summary of actively recruiting clinical trials with CD19-targeted CARs
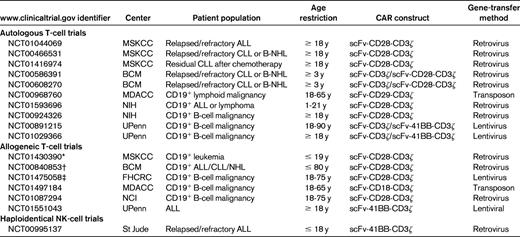
MDACC indicates MD Anderson Cancer Center; NIH, National Institutes of Health; FHCRC, Fred Hutchinson Cancer Research Center; NHL, non-Hodgkin lymphoma; and scFv, single-chain–variable fragment.
*Donor-derived EBV-CTLs.
†Donor-derived Tri-virus CTLs.
‡Donor-derived CD8+ central memory viral specific (EBV or CMV) T cells.
Published results of adoptive therapy with autologous CD19-targeted, CAR-modified T cells
Early reported clinical outcomes from the City of Hope and Baylor College of Medicine (BCM) in patients with low-grade B-cell malignancies treated with CD19-targeted, CAR-modified T cells were disappointing, with patients achieving at best stable disease.65,66 It is notable that the clinical trial designs of these early studies entailed infusion of CD19-targeted, CAR-modified T cells in the absence of prior lymphodepleting or conditioning chemotherapy, a likely clinically relevant variable. The first truly promising clinical outcome with CD19-targeted, CAR-modified autologous T cells was reported by researchers at the National Cancer Institute (NCI). In that case report, a heavily pretreated patient with follicular non-Hodgkin lymphoma received conditioning cyclophosphamide and fludarabine chemotherapy, followed by infusion of T cells modified to express a second-generation CD28-containing CD19-targeted CAR, followed by additional exogenous IL-2 cytokine support.67 The patient achieved a partial response and developed a long-standing B-cell aplasia, a surrogate marker for a persistent anti–CD19-targeted effect mediated by the infused CAR-modified T cells. Soon thereafter, investigators from the University of Pennsylvania (UPenn) published 2 reports detailing a partial response in one patient and long-term molecular complete responses in an additional 2 patients with CLL after conditioning chemotherapy and infusion of T cells modified to express a CD19-targeted, 4-1BB–containing, second-generation CAR.68,69 In addition to impressive antitumor responses, all patients in this cohort exhibited the predicted long-term B-cell aplasia. In similar trials conducted at our institution, treatment with autologous T cells modified to express a CD28-containing second-generation CD19-targeted CAR resulted in a significant radiographic response to disease in 1 of 4 evaluable CLL patients who received prior cyclophosphamide conditioning chemotherapy, with an additional 2 patients achieving stable disease despite rapid tumor progression before therapy.70 In addition, in one patient with relapsed B-ALL, we reported an expected long-term B-cell aplasia after reinduction chemotherapy and genetically modified T-cell infusion before subsequent allo-HSCT. Most recently, investigators at the NCI published the results of a subsequent cohort of 8 patients with low-grade B-cell malignancies.71 That study, which included an analysis of the first published patient after reinfusion of genetically modified T cells, used a CD28-containing second-generation CAR targeted to the CD19 Ag. In these studies, the investigators reported one complete response, one death on study unrelated to T-cell infusion, and partial responses observed in the remaining treated patients. Four of 8 treated patients in this report developed the anticipated B-cell aplasia.
Relevant variables in published CD19-targeted, CAR-modified T-cell clinical trials
Given the increasing number of reported clinical trial results from multiple academic centers, we are now able to speculate on factors that may be relevant to the optimal clinical approach of CAR-modified T-cell therapy. Multiple differences in clinical trial design using genetically modified T cells to treat B-cell malignancies exist, and the optimal clinical trial design using this technology is still to be determined (Figure 2). Specifically, potentially relevant variables between published trials include the design of the CD19-targeted CAR, in vitro T-cell production, the role of prior conditioning chemotherapy, and the roles of tumor burden as well as tumor sensitivity to conditioning therapy at the time of modified T-cell infusion (Table 2).

Variables in clinical trial design. Multiple, potentially clinically relevant variables exist between various published clinical trial outcomes treating patients with CD19-targeted, CAR-modified T cells. There are variables in the methodology of CAR gene transfer (1), the design of the CAR (2), the inclusion or exclusion of prior conditioning chemotherapy (3), whether conditioning chemotherapy may reduce tumor burden (4), and whether additional cytokine support with IL-2 is provided exogenously after modified T-cell infusion (5). Whether one or more of these variables are indeed relevant to ultimate clinical outcomes awaits additional multicenter trials resolving these variables by direct comparison to establish the optimal conditions in which these CAR-modified T cells may induce an optimal clinical response.
Variables in clinical trial design. Multiple, potentially clinically relevant variables exist between various published clinical trial outcomes treating patients with CD19-targeted, CAR-modified T cells. There are variables in the methodology of CAR gene transfer (1), the design of the CAR (2), the inclusion or exclusion of prior conditioning chemotherapy (3), whether conditioning chemotherapy may reduce tumor burden (4), and whether additional cytokine support with IL-2 is provided exogenously after modified T-cell infusion (5). Whether one or more of these variables are indeed relevant to ultimate clinical outcomes awaits additional multicenter trials resolving these variables by direct comparison to establish the optimal conditions in which these CAR-modified T cells may induce an optimal clinical response.

CAR design
Preclinical studies have shown superior in vivo antitumor responses mediated by second- and third-generation CARs (Figure 1) containing T cell–costimulatory signaling domains compared with first-generation CARs lacking costimulatory signaling.58,62,64,72 Consistent with these preclinical studies, elegant studies in patients with low-grade B-cell malignancies infused with a combination of first- and second-generation CAR-modified CD19-targeted T cells demonstrated superior persistence of CD28-containing second-generation CAR-modified T cells compared with first-generation CAR-modified T cells.66 Although it is reasonable, in light of these data and subsequent promising clinical data in patients treated with second-generation CARs,66 to predict the clinical superiority of second-generation over first-generation CAR-modified T cells, direct in vivo comparisons between various second-generation CD19-targeted constructs containing either the CD28 or the 4-1BB signaling domains have not been conducted in patients to date. Both complete tumor responses and B-cell aplasia have been reported in patients treated with T cells modified to express either CD28- or 4-1BB–containing second-generation CARs.67–71 Whether either second-generation costimulatory construct has clinical superiority over the other remains to be evaluated directly in the clinical setting. Also untested in the clinical setting is whether variation of the hybridoma-derived single-chain–variable fragment used in these variable CARs, with the FMC63 MAb being incorporated in the NCI and UPenn CARs and the SJ25C1 MAb incorporated in the MSKCC CAR, plays a relevant role in antitumor efficacy and subsequent clinical outcomes (Table 2).
T-cell production
Gene-transfer methodology also varies between centers: either gamma-retroviral vectors or lentiviral vectors are used by most centers, but some use electroporation or transposon technology.74–76 Given the time in culture required for T-cell production using electroporation, this approach has largely been abandoned.76 Published studies lack direct comparisons between alternative gene-transfer approaches, leaving open the question of whether there are clinically meaningful differences between gamma-retroviral-, lentiviral-, and transposon-mediated gene transfer. However, given the published data, it is worthwhile to note that clinical CRs and resultant B-cell aplasia were achieved in the setting of both gamma-retroviral67,70,71 and lentiviral68,69 gene-modified, CD19-targeted, CAR-modified T-cell therapies. In addition, the concern regarding the risk of insertional oncogenesis after gene transfer in the T cell is unknown, but recent studies have demonstrated a decade-long safety using gamma-retroviral vectors.40,76,77 Furthermore, the relative risk of insertional oncogenesis between the various gene-transfer methodologies has yet to be determined.
Prior conditioning chemotherapy
Conditioning chemotherapy, also termed lymphodepleting therapy, is a well-recognized and requisite adjunct to enhancing antitumor efficacy in the setting of autologous tumor-infiltrating lymphocyte therapy for patients with melanoma.79 Based on the currently published clinical outcomes from patients treated with CD19-targeted CAR+ T cells, this paradigm appears to be relevant to the field of CAR-modified T-cell adoptive therapy. Specifically, in 2 initial published clinical trials using CD19-targeted, CAR-modified T cells patients treated in the absence of prior conditioning chemotherapy achieved at best modest clinical responses.65,66 In contrast, unique trials at our institution allowed for the comparison between an initial cohort of patients treated with CAR-modified T cells in the absence of prior conditioning chemotherapy followed by a similar cohort treated with an even lower dose of T cells after prior cyclophosphamide chemotherapy. In these studies, we demonstrated both improved persistence of modified T cells and improved clinical outcomes in the cohort receiving prior conditioning chemotherapy.70 Similarly, studies from both the NCI and UPenn included prior chemotherapy conditioning of patients, which resulted in a marked clinical benefits.67–69,71 Comparisons of the clinical outcomes from these studies support the use of prior conditioning therapy to achieve the optimal clinical outcomes after CAR-modified T-cell therapy.
Tumor burden and tumor sensitivity to conditioning chemotherapy
Analysis of CD19-targeted CAR+ T-cell persistence in treated patients with either CLL or B-ALL at Memorial Sloan-Kettering Cancer Center (MSKCC) demonstrated an inverse correlation between tumor burden and detectable infused modified T cells over time.70 This finding, in combination with our reported clinical outcomes, is consistent with an enhanced anti-CD19 efficacy mediated by CAR-modified T cells in the setting of lower tumor burden. In light of this finding, there remains the additional question of what role potential tumor sensitivity to conditioning chemotherapy plays in the ultimate clinical outcomes of patients treated with CD19-targeted, CAR-modified T cells. Specifically, in our studies, patients were conditioned with cyclophosphamide before genetically modified T-cell infusion. In our cohort of treated CLL patients, patients had received one or more prior chemotherapy regimens containing cyclophosphamide. Patients with tumors already refractory to cyclophosphamide treated on our protocols achieved no additional reduction in tumor burden mediated by this conditioning regimen before infusion with modified T cells.70 In contrast, CLL patients treated on the UPenn protocol were naive to conditioning chemotherapies with standard second-line CLL agents, including bendamustine and pentostatin.68,69 Similarly, the initial reported patient at the NCI was treated with a combination conditioning regimen of cyclophosphamide and fludarabine, the latter of which the patient had not received previously.67 These results, at the very least, suggest a potential correlation between clinical outcomes and potential tumor sensitivity to prior conditioning chemotherapies. Unfortunately, the more recently published clinical data from the NCI failed to report prior patient chemotherapy regimens to allow for further correlation between genetically modified T-cell antitumor efficacy and tumor sensitivity with respect to the prior conditioning chemotherapy.71
Additional currently ongoing clinical trials of B-cell malignancies using CD19-targeted, CAR-modified T cells
Given the promising published clinical outcomes of patients with B-cell malignancies treated with CD19-targeted, CAR-modified T cells to date, there has been a rapid expansion of additional trials at multiple centers using this CD19-targeted CAR technology (Table 1). Specifically, at our institution, we have recently opened an additional clinical trial treating CLL patients in the upfront setting with CD19-targeted, CAR-modified T cells as a consolidation after initial therapy with pentostatin, cyclophosphamide, and rituximab–based chemotherapy. Additional CD19-targeted CAR clinical trials are currently open using transposon gene-transfer technology73 and CAR-modified T cells generated in vitro from previously selected T cells with a central memory phenotype, which may enhance subsequent in vivo persistence79 (Table 1).
Several centers, including MSKCC, have transferred this technology from the autologous to the allogeneic setting. Specifically, at our institution, we have initiated enrollment and treatment of pediatric patients with B-ALL who have relapsed after allo-HSCT with donor-derived EBV-specific T cells modified to express our CD28-containing second-generation CAR targeted to the CD19 Ag. The rationale of this approach is based on the reported safety and minimal GVHD-mediated toxicity that these enriched EBV-specific allogeneic T cells have in patients with EBV-LPD. Our institution has recently reported on the safety of infused donor EBV-CTLs for patient with EBV-LPD with the development of GVHD.39 Our aim is to provide a GVL effect through the anti-CD19 CAR in the absence of a GVHD despite the allogeneic nature of these T cells. Similar studies using CD19-specific, CAR-modified viral specific T cells with specificity against 3 viruses (EBV, CMV, and adenovirus)80 are currently being conducted by investigators at BCM (Table 1).
Genetically engineered adoptive therapy of B-cell malignancies is not solely restricted to T cells. In fact, based on the clinical potential of NK cells as antileukemic immune effectors in the haploidentical setting, investigators have reported promising preclinical studies using CD19-targeted, CAR-modified NK cells.81 Based on these promising preclinical data, investigators at St Jude Research Hospital have opened a clinical trial treating patients with haploidentical NK cells also modified to express a second-generation 4-1BB–containing, CD19-targeted CAR (Table 1).
Additional target Ags for genetically modified immune effectors in the adoptive immune cell therapy of leukemias
The restricted nature of the CD19 Ag on normal B cells and most B-cell malignancies has resulted in this target receiving the majority of attention in both preclinical and clinical investigations for adoptive T-cell therapy in leukemia. However, additional target Ags with the potential to broaden the application of this approach to other hematologic malignancies are under investigation. Specifically, clinical trials using CAR-modified T cells targeted to the CD20 Ag have demonstrated safety and some promising initial clinical outcomes.65,75 Additional targets for CAR-modified T cells include the receptor tyrosine kinase-like orphan receptor 1 (ROR1) or kappa-light chain of human Ig, which expands the application of this therapy for hematologic malignancies.82,83 Furthermore, this adoptive T-cell approach may be extrapolated to myeloid malignancies based on promising preclinical data using CAR-modified immune effector cells targeted to the CD33 Ag and the Lewis Y Ag,84–86 as well as TCR-modified T cells targeted to the WT-1 Ag and the hyaluronan-mediated motility receptor (HMMR/Rhamm).87,88 With the exception of anti-kappa chain CAR-modified T cells, it is unclear if these latter Ags will be targeted through adoptive T-cell therapies in future clinical trials.
Further modification of tumor targeted T cells: The next generation
To date, CAR-modified T cells have demonstrated promising initial clinical responses; however, most patients who have been treated with CD19-targeted T cells ultimately develop progressive disease and succumb to their disease. We and others have developed additional genetic approaches to enhance the in vivo antitumor efficacy of CAR-modified T cells. Specifically, we have demonstrated the additional genetic modification of CAR-modified T cells to express proproliferative T cell–costimulatory ligands (4-1BBL)89 or proinflammatory cytokines (IL-12),90 resulting in “armored” fourth-generation CAR-modified T cells (Figure 1). These cells have shown enhanced in vivo antitumor efficacy in preclinical tumor models compared with T cells modified to express the tumor-targeted CAR alone. Based on the very promising preclinical outcomes of these studies, we anticipate translating these more potent tumor-targeted T cells to the clinical setting as the next generation of CAR-modified T-cell trials.
Conclusions
Under ideal conditions, immune effectors may mediate a GVL effect, which in turn may result in an enhanced clinical response in the allogeneic setting for patients with hematologic malignancies. Unfortunately, this GVL effect in the setting of most hematologic malignancies is at best modest, with many patients experiencing relapsed disease despite allo-HSCT and DLI therapy. Gene therapy of immune effectors to target these cells to Ags expressed on tumor cells have demonstrated promising clinical responses in the autologous setting. However, despite promising but anecdotal early clinical trial results in the setting of T cells targeted to the B-cell CD19 Ag, significant variables in clinical trial design make it difficult to determine the optimal clinical setting in which to apply this novel technology. Variables in CAR design, gene-transfer technology, and prior conditioning therapies need to be addressed directly in the clinical setting to answer these questions. Given the increasing number of clinical trials using CD19-targeted, CAR-modified T cells currently open at multiple centers, these questions are doomed to remain largely unanswered through direct clinical comparisons in the absence of collaborative trials between centers. Our institution, in collaboration with investigators at UPenn and the Children's Hospital of Philadelphia through a National Institutes of Health Special Translational Research Acceleration Project (STRAP) award, are currently addressing several of these variables directly by harmonizing our respective clinical protocols. Through this collaboration, patients with B-cell malignancies will be treated with equal numbers of autologous T cells modified with the gamma-retroviral gene transfer of the MSKCC CD28 containing second-generation, CD19-targeted CAR and the UPenn lentiviral gene transfer of the 4-1BB–containing second-generation CD19-targeted CAR. These studies are designed to address the questions of both optimal second-generation CAR design (CD28 versus 4-1BB containing CARs) and gene-transfer technology (gamma-retroviral versus lentiviral vectors) directly. However, these studies do not directly address additional questions regarding optimal conditioning therapy and the relationship between CAR-modified T-cell antitumor efficacy and tumor sensitivity to conditioning chemotherapy. We hope that this initial collaborative effort will serve as a template to other investigators in this field and spur additional collaborative trials to further compare additional variables, not only with respect to targeting tumors expressing the CD19 Ag, but also with respect to clinical trials with CAR-modified T cells targeting additional promising Ags expressed on a wider array of hematologic and solid tumor malignancies.
Disclosures
Conflict-of-interest disclosure: The authors declare no competing financial interests. Off-label drug use: None disclosed.
Acknowledgments
The authors thank Hollie Pegram for designing the figures; Michel Sadelain and Joe Olechnowicz for reviewing the manuscript; and Isabelle Riviere, Nancy Kernan, Jae Park, Marco Davila, Xiuyan Wang, and the entire CTCEF team for their efforts on behalf of the patients.
Correspondence
Renier Brentjens, MD, PhD, Associate Member, Leukemia Service, Department of Medicine, Memorial Sloan-Kettering Cancer Center, 1275 York Ave, New York, NY 10065; Phone: 212-639-7053; Fax: 212-717-3569; e-mail: brentjer@mskcc.org.
