
Abstract
Pregnancy poses a unique challenge to patients with sickle cell disease and β-thalassemia, who often have exacerbations of hemolysis or anemia during the gestational period, experience higher rates of obstetric and fetal complications, and may have distinct underlying comorbidities related to vasculopathy and iron overload that can endanger maternal health. Optimal management of pregnant women with hemoglobinopathies requires both an understanding of the physiologic demands of pregnancy and the pathophysiology of disease-specific complications of inherited blood disorders. A multidisciplinary team of expert hematologists and high-risk obstetricians is therefore essential to ensuring appropriate antenatal maternal screening, adequate fetal surveillance, and early recognition of complications. Fortunately, with integrated and targeted care, most women with sickle cell disease and β-thalassemia can achieve successful pregnancy outcomes.
Introduction
Sickle cell disease (SCD) and β-thalassemia are autosomal-recessive disorders of the β-globin gene that result in severe anemia. In SCD, a point mutation of the β-globin gene results in the production of an abnormal hemoglobin (HbS) that polymerizes in its deoxygenated state and forms sickle-shaped RBCs.1 Acute complications of the disease such as pain crisis and acute chest syndrome (ACS) are due in part to vasoocclusion from direct entrapment of the deformed RBCs in the microcirculation, whereas chronic sequelae such as pulmonary hypertension and leg ulceration result from long-term toxic effects from intravascular hemolysis and vascular dysfunction.2 In β-thalassemia, decreased or absent synthesis of the β-globin gene results in ineffective erythropoiesis and significant anemia secondary to both underproduction and hemolysis from unstable α-globin inclusions.3 Although the clinical manifestations of β-thalassemia depend on modifying genetic factors, morbidity associated with the disease is common. Extensive extramedullary hematopoiesis in β-thalassemia patients can lead to early complications such as skeletal deformities and splenomegaly, and iron overload due to increased gut iron absorption and transfusion therapy results in late organ dysfunctions such as endocrine abnormalities and cardiac failure.3 Approximately 90 000 people are affected by SCD in the United States alone,4 and although the prevalence of β-thalassemia major (TM) in the United States is estimated at only 1000,5 β-thalassemia subtypes, including both β-TM and β-thalassemia intermedia (TI), affect a considerable proportion of births worldwide, especially in areas with high carrier frequency such as India and southeast Asia.6
Improvements in the overall management of hemoglobinopathies have allowed affected persons to enjoy an improved quality of life and increased life expectancy. However, pregnancy can lead to maternal and fetal complications in SCD and β-thalassemia patients, even in those who are otherwise asymptomatic. Because research regarding the optimal management of pregnant patients with inherited hemoglobin disorders is sparse, specialty care with trained hematologists and maternal-fetal specialists familiar with hemoglobinopathies is essential for the treatment of these disorders and their complications during gestation. Clinically severe α-thalassemia syndromes such as hemoglobin H disease variants are rare6 and the management of pregnant women with α-thalassemia and a prenatal diagnosis of hydrops fetalis are beyond the scope of this review.
Maternal and fetal complications
SCD
Pregnancy results in a myriad of physiologic changes that can interact adversely with sickle hemoglobin to promote abnormal RBC sickling. Oxygen demand during pregnancy increases to support the metabolic requirements of the developing placenta and fetus.7 Although normal compensatory mechanisms such as an increased blood volume often result in adequate maternal and fetal oxygen delivery in a healthy woman, the need for an increased plasma volume relative to erythrocyte production can significantly worsen anemia in patients who already have severely low hemoglobin levels and shortened erythrocyte survival. Furthermore, the maternal oxygen reserve may be compromised during pregnancy secondary to the increased oxygen consumption and decreased functional residual capacity during gestation,7 which may predispose SCD patients to hypoxemia and subsequent exacerbation of sickling. Therefore, maternal and fetal morbidity in SCD stems from an increase in both pregnancy-related complications relating to the degree of anemia and impaired oxygen delivery and complications associated with SCD itself.
Although there is a general consensus that the overall risk of complications during pregnancy for patients with SCD is increased, frequency estimates for specific obstetric outcomes have varied widely between studies. The largest cohort study from the Cooperative Study of Sickle Cell Disease found generally favorable outcomes in 445 pregnancies, 286 of which progressed through to delivery.8 In that study, of the pregnancies that were not electively terminated, 89.3% progressed to live births, with a mean gestational age of 37.7 weeks. In addition, mortality was low, with only 2 pregnancies resulting in maternal death, and rates for miscarriage (6.5%) and non–SCD-related obstetric complications were not found to be increased in women with SCD compared with the reported rates for the general African-American population. However, the risk of small for gestational age babies did appear to be exceedingly high in SCD patients, with 77% of mothers with hemoglobin SS disease delivering infants with birth weights below the 50th percentile. Another longitudinal observational study performed in Jamaica of 94 pregnancies in SS patients found similarly low non–SCD-related obstetric complications, but noted an increase in spontaneous abortions (36%) compared with 157 pregnancies in controls (10%).9 In addition, increased maternal mortality in SCD patients was noted in the Jamaican study, but was based on only 2 maternal deaths. Studies performed in patients with SC or Sβ+ genotypes have demonstrated a high risk of intrauterine growth restriction (IUGR), but subsequent low birth weight has not been found to be more common in patients with sickle hemoglobin variants compared with control populations.8,10,11
Because of the rarity of SCD and the limited number of pregnancies available in observational studies of SCD patients, a few large database studies have been published recently to provide pregnancy outcome data on a larger scale. The most comprehensive of these evaluations was performed in a study of nearly 18 000 deliveries to SCD mothers compared with controls using a national inpatient database, and the results are summarized in Table 1.12 In that study, infectious sequelae, including systemic inflammatory response syndrome, sepsis, urinary tract infections, and pneumonia, were significantly more common in pregnant women with SCD, as were thrombotic complications such as cerebral vein and deep venous thrombosis. SCD patients were also more prone to pregnancy-specific complications, including preeclampsia, eclampsia, placental abruption, and antepartum bleeding. Consistent with the prior cohort reports, IUGR was seen more often in patients with SCD, and rates of maternal mortality were significantly higher in women with SCD than in controls (72.4 deaths vs 12.7 per 100 000, respectively). The increased thrombotic risk during pregnancy in patients with SCD was confirmed in a study of venous thromboembolism (VTE) risk factors using the same national database, with an odds ratio (OR) of 6.7 for development of VTE in SCD patients compared with controls.13 Similarly, the high maternal mortality among SCD patients was also demonstrated in a recent Jamaican death registry study, although the estimated rates of maternal death were nearly 10 times higher than those reported for the United States (719 deaths per 100 000 deliveries), which the investigators noted may be related to limited access to intensive and specialty care.14
Complications of pregnancy among women with SCD
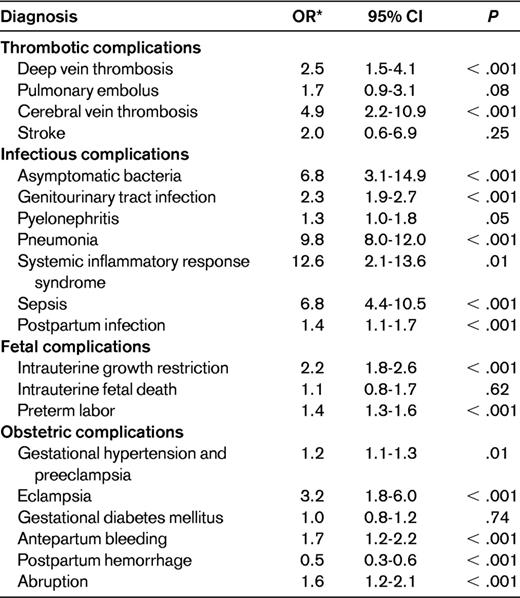
Adapted from Villers et al.12
CI indicates confidence interval.
*ORs are listed for women with SCD compared with women without SCD.
β-thalassemia
In contrast to SCD, data regarding maternal and fetal outcomes in β-thalassemia are extremely scarce, both because a substantial proportion of affected patients live in developing countries6 and because hypogonadotrophic hypogonadism secondary to transfusional iron overload can occur in greater than 60% of β-TM patients,15 necessitating fertility treatment for successful conception in a majority of women. Nonetheless, the few available case series do provide insights into the potential complications associated with pregnancy in this population and raise awareness into the potential management issues of pregnancy in thalassemia patients.
The mainstay of treatment for β-TM includes transfusion therapy, which is often instituted in early childhood, coupled with aggressive iron chelation therapy to prevent iron accumulation. In patients with iron-overload, cardiac involvement and arrhythmia are a major cause of early morbidity and mortality3 and an exceedingly high maternal mortality rate has been reported in patients with β-TM with known cardiac iron deposition. In one study, 2 (6.9%) of 29 pregnancies resulted in maternal death in the postpartum period, both of which were in women with preexisting cardiac dysfunction.16 Another study from Greece also reported deaths due to heart failure during the second trimester of gestation in women with poorly managed cardiac hemosiderosis.17 Several hemodynamic changes during pregnancy affect cardiac function and are known to cause an increase in maternal cardiac stress in the gestational period. An increase in blood volume, decrease in systemic vascular resistance, and augmentation of cardiac output occurs in all normal pregnancies to match maternal-fetal demands.7 In one study of echocardiography in pregnant β-thalassemia patients without cardiac impairment, an appropriate increase in cardiac output was observed secondary to an increase in resting heart rate and left ventricular mass.17 Although cardiac function did appear to return to pre-pregnancy levels after delivery, a subset of β-TM patients was noted to have a late decline in cardiac reserve in the years after gestation; however, the long-term outcome for these patients is unknown.
More recent reports in β-TM patients without cardiac impairment and treated with aggressive pre-pregnancy chelation have generally observed favorable outcomes. One of the largest modern studies of 58 pregnancies in β-TM mothers found that impaired glucose tolerance and gestational diabetes were frequent complications, occurring in 15% of patients.18 Worsening of anemia during pregnancy also led to an increase in transfusion requirements of a median of 25 mL/kg/y and a subsequent 60% increase in baseline ferritin. Fetal complications such as IUGR and premature delivery did appear to be increased in β-TM patients, occurring in nearly 40% of infants; however, because many of these women required fertility treatments, twin pregnancies accounted for a major number of these births.
In β-TI, the most significant adverse maternal outcome appeared to be thrombotic events, which occurred in 7.2% of 83 pregnancies in one series.19 Development of anemia resulted in a need for transfusion in 60%-80% of β-TI patients, 30% of whom had never had a transfusion before pregnancy.18,19 IUGR and prematurity rates were also noted to be high among β-TI mothers, occurring in 20% and 30% of infants, respectively.19
Antepartum considerations
Genetic testing
Optimal obstetric management of patients with hemoglobinopathies requires an accurate assessment of the underlying hemoglobin genotype to accurately anticipate potential complications. Whereas the majority of patients with sickle cell anemia (SS/Sβ0) or β-TM will have established regular hematology care far before conception, female patients with SC, Sβ+, or β-TI may be undiagnosed, asymptomatic, or have infrequent need for specialty care; therefore, all patients with unclear disease type or a suspicion for hemoglobinopathy should undergo diagnostic testing with complete blood count, hemoglobin analysis, and quantification of hemoglobin A2 levels early in their pregnancy. After confirming pregnancy or a hemoglobin disorder, questions regarding the probability of delivering a child with a hemoglobinopathy often arise. Genetic testing should be offered to the partner to determine carrier status, if unknown, to determine the risk of delivering an affected child; in fact, national efforts for antenatal screening have been adopted for high-risk ethnic groups in some countries.20 If the partner does carry an abnormal gene, counseling should be aimed at discussing the 50% risk of having an affected child, but reassuring the couple that fetal defects and demise secondary to the disease are not common because the β-globin gene is not expressed significantly until well after birth.21
Medication modifications
A comprehensive review of medications early in pregnancy is warranted to avoid potential teratogenic effects of disease-specific therapies. Hydroxyurea remains a principle therapy in sickle cell anemia patients with frequent acute symptoms or evidence of end-organ damage,22 and female patients may inadvertently become pregnant while taking the drug. Experimental animal models, including both rats and primates, have demonstrated fetal death and birth defects such as brain, cardiac, and skeletal abnormalities in embryos exposed to hydroxyurea in utero.23 Only a few studies have described the effects of hydroxyurea in human subjects and have been inconclusive secondary to low numbers of included patients. In one case series of 31 women using hydroxyurea for various indications, no fetal malformations were noted, but 3 pregnancies resulted in in utero death or spontaneous abortion.24 These data have led the National Toxicology Program expert panel to recommend against the use of hydroxyurea during pregnancy.22 Therefore, although consideration of continuing the medication during pregnancy has been left to specialists, our practice is to recommend against becoming pregnant while on hydroxyurea and to universally discontinue its use at the first suspicion or confirmation of pregnancy. For male patients, there are no data that hydroxyurea use at the time of conception is associated with an increased risk of birth defects. However, animal models suggest that the drug does have an adverse effect on spermatogenesis,22 so short-term cessation of hydroxyurea may be required in male patients whose partners are trying to conceive.
Many patients with SCD will be prescribed chronic narcotic treatment before conception. In general, the dose of long-acting analgesia should not be modified in pregnancy because narcotics are often required to provide effective pain relief during gestation and have not been associated with congenital defects.25 Furthermore, a recent meta-analysis of methadone treatment during pregnancy demonstrated that the severity of neonatal abstinence syndrome (NAS) was not dependent on the dose of medication.26 Although all chronic narcotic regimens are associated with a risk of NAS, a recent study evaluating pregnant women who were prescribed various prescription narcotics for pain demonstrated an incidence of NAS of only 5.6%, which is significantly lower than the rates reported for patients on methadone maintenance for addiction.27 Some mothers, however, may request to reduce their dose of analgesia while pregnant, and the decision regarding dose modification should involve a discussion about the potential risks and benefits. If the dose is lowered or the medication discontinued altogether, acute withdrawal should be avoided because abrupt cessation can be associated with adverse fetal outcomes, including preterm delivery and death.25
In β-TM predominantly, chelation therapy with either injectable (deferoxamine) or oral (deferiprone or deferasirox) formulations is used to treat transfusional iron overload. Of these drugs, deferoxamine has been the most extensively evaluated in pregnancy, in part because it is the oldest. Animal studies have noted skeletal abnormalities in rats exposed to deferoxamine early in gestation, although the teratogenic effect was greatest at doses that also caused maternal toxicity.28 Numerous subsequent case series, the largest of which followed 32 women with β-TM, have demonstrated favorable fetal outcomes in patients treated with deferoxamine chelation during the second and third trimesters.29 Given these findings, deferoxamine should be avoided during the first trimester, but may be considered in the second and third trimesters in patients with a strong indication for treatment.3,29 In addition, the drug should be stopped promptly if any maternal toxicity develops, and some experts also recommend using only the subcutaneous formulation of deferoxamine during pregnancy to minimize hemodynamic toxicity.3 With respect to the oral chelators, experimental models have shown that placental transfer of deferasirox is minimal in rats,30 but fetal outcome data in humans are limited to case reports. Deferiprone has only recently been improved in the United States, and its effect on developing embryos is unknown. As a result, oral formulations should be not be used during pregnancy, and if chelation is necessary during gestation, substitution with deferoxamine is indicated. In addition, prenatal vitamins with folate should be advocated for all patients with hemoglobinopathies, but care should be taken to avoid preparations with iron supplementation in females with iron overload.21
Assessment of comorbidities
Patients with hemoglobinopathies often have preexisting conditions that require frequent monitoring during pregnancy. Therefore, all patients with SCD or thalassemia should be screened for relevant comorbidities in early gestation so that consultation with cardiac or pulmonary specialists can be initiated. Pulmonary arterial hypertension is associated with an extremely high mortality rate during pregnancy,31 and has been increasingly recognized as a complication of both β-thalassemia and SCD.2,32 Given the substantial maternal risks, screening for pulmonary hypertension is warranted for patients who have not undergone echocardiographic testing within the year before pregnancy. Testing should be initiated as early in pregnancy as possible, because physiologic changes during gestation can result in inaccurate echocardiographic measurements.31 Although tricuspid regurgitant jet velocity ≥ 2.5 m/s has been shown to be a sensitive screening tool for pulmonary hypertension in SCD, it is associated with a high false-positive rate and right heart catheterization is required to confirm the diagnosis.33 Therefore, any female patient with an elevated tricuspid regurgitant jet velocity should be referred to a pulmonary clinic for evaluation and monitoring during pregnancy.
In patients with β-TM, the high maternal mortality observed in women with underlying cardiac impairment has led to strong recommendations for women with cardiac iron overload to avoid pregnancy.16,17 However, pregnancy may still occur in patients with occult or overt cardiac dysfunction and active surveillance must be instituted early to prevent maternal morbidity. Because preclinical myocardial siderosis in β-TM is common even in those on aggressive chelation protocols, all patients who have not received annual dedicated cardiac imaging should undergo echocardiography to assess left ventricular function at the first antenatal visit. Repeat screening for cardiac dysfunction may also be warranted for patients who develop cardiac symptoms during gestation, especially those with continued transfusion requirements, because the hemodynamic stress of pregnancy may exacerbate or unmask occult myocardial disease.17 Given the significant potential maternal risks, referral to a cardiologist should be undertaken at the first sign of cardiac impairment and consideration should be given to initiating chelation therapy as early as the second trimester.29
Iron overload is also associated with a high incidence of endocrine abnormalities, including diabetes and hypothyroidism, in β-TM patients.15 Maternal hyperglycemia is associated with a host of adverse pregnancy outcomes, including fetal macrosomia, birth injury, neonatal intensive care, preeclampsia, and premature delivery, and universal screening for diabetes is generally performed at 24-32 weeks of gestation.34 Because β-TM patients may have impaired glucose tolerance or undiagnosed diabetes before conception,18 pregnant women with β-TM should be screened at their first prenatal visit to allow for early intervention. Similarly, severe maternal hypothyroidism has been associated with poor fetal outcomes such as miscarriage and preterm delivery.35 Thyroid screening should therefore be performed in β-TM patients in the first trimester, but care should be taken to use trimester-specific reference ranges for thyroid-stimulating hormone, because baseline levels of this hormone are depressed throughout pregnancy.
Blood pressure monitoring
Baseline blood pressure in patients with SCD is commonly lower than that in the general population21 ; however, pregnancy-related hypertensive disorders in SCD are remarkably common. In a large database study, SCD conferred a 1.2 OR of gestational hypertension and preeclampsia compared with controls, and the risk of eclampsia was substantially higher, with an OR of 3.2.12 At our institution, we have observed preeclampsia rates greater than 20% in our pregnant patients with SCD. Given the disproportionate risk of SCD patients developing severe sequelae of hypertension, such as eclampsia and seizure, increases in systemic blood pressure or evidence of proteinuria after 20 weeks should be monitored aggressively and may require inpatient evaluation.36 There is no consensus about the optimal threshold for initiation of antihypertensives in pregnant women, and decisions regarding treatment should be based both on the degree of hypertension and presence of end-organ damage.
Fetal surveillance
Chronic anemia and microvascular sickling can result in low placental weights and impaired blood flow in pregnant women with SCD, and decreased umbilical flow in the third trimester is correlated significantly with low neonatal birth weights.37,38 Although studies verifying placental insufficiency have not been performed in β-thalassemia, the high rates of IUGR in select β-thalassemia patients suggests that ureteroplacental hypoxia may be a key feature of this disease.16 Current obstetric guidelines advocate the initiation of fetal monitoring in high-risk patients at 32-34 weeks, and surveillance can be instituted as early as 26 weeks if the mother has additional underlying comorbidities.39 In addition to fetal nonstress and biophysical profile testing, umbilical artery Doppler after 28 weeks has been shown to improve fetal outcomes in high-risk pregnancies because of its ability to identify infants who would benefit from early delivery.40 Therefore, to reduce perinatal mortality, routine use of Doppler ultrasonography is recommended for all patients with underlying hemoglobinopathies.40
Management of disease-specific complications
Anemia/transfusion
The need for transfusion during pregnancy secondary to either acute severe anemia or obstetric emergency is common in patients with SCD, occurring in 50% of SS and Sβ patients and up to 30% of patients with SC disease.10,11 Empiric transfusion or erythrocytapheresis has been proposed as a method to decrease adverse maternal and fetal outcomes in SCD, but studies evaluating its efficacy have demonstrated conflicting results.11,41–44 A single randomized controlled trial evaluated the benefit of an aggressive transfusion protocol to maintain a hemoglobin concentration between 10 and 11 g/dL in pregnant women with SS disease, and found that patients who received prophylactic transfusions did not experience a significant reduction in obstetric or fetal outcomes compared with women treated with transfusion for medical or obstetric complications or hemoglobin levels less than 6 g/dL.11 Subsequent retrospective cohort studies evaluating the efficacy of prophylactic simple transfusion also failed to show a substantial decrease in obstetric and fetal complications.41,42 Conversely, empiric erythrocytapheresis has been associated with a reduction in maternal complications such as ACS and adverse fetal outcomes, including IUGR, low birth weight, and perinatal mortality.43,44 Furthermore, the frequency of vasoocclusive crisis (VOC) did appear to be decreased significantly in pregnant SCD patients in all of the simple and exchange transfusion studies.11,41–44 However, differences in the trimester in which prophylactic protocols were initiated and inconsistencies in transfusion goals limit the interpretation of these results, and at this point, empiric transfusions are not currently the standard of care for SCD patients. Although specific simple transfusion goals have not been established, pregnant SCD patients should be transfused for hemoglobin values < 6 g/dL in accordance with general obstetric guidelines, because this threshold has been associated with abnormal fetal oxygenation and fetal death in non-SCD populations.45 Transfusion at a higher threshold may be warranted for management of SCD-specific indications such as ACS or obstetric complications. In our practice, we offer chronic simple or exchange transfusion regimens to our patients who develop a severe SCD-related event such as recurrent VOC, ACS, or organ failure during pregnancy, as well as to those who had been prescribed hydroxyurea for SCD complications before conception, because the risk of recurrent events remains high throughout the gestational period.
In β-TM, preconception transfusion goals (> 9-10 g/dL) are generally continued throughout pregnancy to optimize fetal and maternal well-being,16,18 and exacerbation of anemia requiring an increase in transfusion requirement in β-TM patients occurs nearly universally.18 Transfusion practices in pregnant women with β-TI, however, are highly variable, with some centers advocating transfusion in all patients with a hemoglobin < 10 g/dL and others reserving transfusion for only those patients with severe anemia or evidence of IUGR.18,19 However, no studies have evaluated obstetric outcomes based on hemoglobin goals in β-TI, and therefore the decision to transfuse should be individualized depending on maternal and fetal indications.
Alloimmunization
Alloimmunization can occur in transfused patients with hemoglobinopathies, and high rates of alloantibody production have been noted in both SCD and β-thalassemia patients.46,47 Pregnancy itself is a risk factor for alloantibody development secondary to exposure to foreign fetal antigens. Transfusion-related alloimmunization may also occur more frequently during gestation, although epidemiologic data regarding this phenomenon are not clear.48 Nonetheless, in studies of pregnant SCD patients receiving transfusions, up to 30% will have detectable alloantibodies before conception,43 with another 5%-20% developing new Abs during pregnancy.41,49 Similarly, in β-thalassemia, rates of alloimmunization are exceedingly high, ranging from 5%-30% depending on the country in which the study was performed.47 Alloimmunization can lead to significant morbidity during pregnancy, and both severe delayed hemolytic transfusion reactions and hemolytic disease of the newborn have been described in hemoglobinopathy patients.18,19,41,49 Therefore, a comprehensive history of prior alloimmunization should be elicited from all pregnant patients with inherited blood disorders, and consideration should be given to performing alloantibody screening in early pregnancy to document existing titers. Patients who are heavily alloimmunized and require RBC transfusion should receive extended phenotypically matched products. In addition, because minor antigen matching has been found to be effective in reducing the rates of alloimmunization in SCD and thalassemia patients46,47 without a history of alloimmunization, limited phenotypic matching should be considered whenever possible.
Acute VOC
In females with SCD, painful episodes requiring hospitalization during pregnancy are common, with the incidence of VOC ranging from 20%-30% in pregnant women with the SC genotype to nearly 50% in SS, Sβ0, and Sβ+ patients.8,10 Although the Cooperative Study of Sickle Cell Disease did not find an increased frequency of VOC during pregnancy,8 inpatient management of pain crises is still required by many pregnant SCD patients. Management of acute VOC during pregnancy involves traditional paradigms of providing IV fluids and supplemental oxygen to decrease hypoxia. Intermittent short-acting opiates can be safe during pregnancy and should be dosed to provide adequate pain relief.25 In addition, because infectious complications occur at a high rate in pregnant SCD patients,12 a comprehensive evaluation with routine laboratory testing, blood and urine cultures, and chest X-ray with appropriate fetus shielding is warranted to allow for early initiation of antibiotics. ACS during pregnancy can occur in up to 20% of patients9 ; therefore, any new fever, oxygen requirement, or infiltrate should prompt an immediate assessment for the need for treatment with simple or exchange transfusion.
VTE
Patients with chronic hemolytic anemia syndromes have an overall increased risk of VTE, and the rate in SCD patients during pregnancy has been verified to be higher than that in controls.12,13 Death secondary to VTE has been also been described in SCD, one of which occurred during the second trimester.8,9 The prevalence of VTE has also been noted to be high in β-thalassemia patients, especially those with β-TI and those treated with splenectomy, and thrombosis has been found to complicate a significant proportion of pregnancies in β-TI patients.19 Currently, there are no prospective trials evaluating the benefit of antepartum or postpartum anticoagulation in patients with hemoglobinopathies. In Italy, favorable outcomes have been achieved with prophylaxis with antenatal aspirin followed by postpartum low-molecular-weight heparin in β-thalassemia patients, but the number of patients treated was small.18 For now, anticoagulation should only be considered in those with prior VTE, and care should be taken to elicit a complete history of thrombotic events to identify those patients who could benefit from treatment.
Delivery/postpartum considerations
Delivery
Pregnancy in women with hemoglobinopathies should be allowed to progress to term when possible, because planned preterm delivery in the absence of fetal or maternal indications has not been demonstrated to improve outcomes even in high-risk pregnancies.40 Rates of cesarean section in β-TM patients approaches 80%, with cephalopelvic disproportion being the most common indication for surgical delivery.16 Complications of iron overload, including short stature secondary to delayed pubertal growth and fetal macrosomia in the setting of diabetes, likely contribute to this problem in β-TM mothers. In SCD, however, vaginal delivery is often possible and should be favored.21 General anesthesia should be avoided in SCD given the increased risk of complications such as ACS and should only be considered if exchange or adequate simple transfusion can be planned appropriately.
Breastfeeding
All patients with hemoglobinopathies should be encouraged to breastfeed, but the duration of breastfeeding should be tailored based on the need to resume medications. Hydroxyurea is excreted in breast milk,22 so we do not recommend restarting it until after the patient has ceased nursing. Chronic narcotics can be continued during breastfeeding,25 but because opioids are also excreted in breast milk, decisions regarding long-term nursing must be individualized based on the dose and duration of medication required. In mothers with β-TM, chelation treatment may need to be resumed in the postpartum period. Minimal levels of chelation drugs are secreted into mother's milk,30 so the risks and benefits must be weighed before resuming the medication in breastfeeding women.
Conclusion
With improved life expectancy in the hemoglobinopathies, many women with these disorders are now choosing to become pregnant. Although numerous complications can occur, vigilant monitoring by both experienced obstetricians and hematologists can lead to successful pregnancy outcomes.
Disclosures
Conflict-of-interest disclosure: S.L. has been on the board of directors or an advisory committee for HemaQuest and has received research funding from the National Heart, Lung, and Blood Institute (National Institutes of Health, Bethesda, MD). R.P.N. declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Sophie Lanzkron, MD, MHS, Sickle Cell Center for Adults at Johns Hopkins, 1830 E Monument St, Suite 7300, Baltimore, MD 21205; Phone: 410-502-7770; Fax: 410-614-8601; e-mail: slanzkr@jhmi.edu.
