
Abstract
A 10-year-old male patient with homozygous sickle cell disease presents for a follow-up clinic visit after a recent hospitalization for a painful vasoocclusive event. His parents mention that in the past year he has had 4 hospitalizations for vasoocclusive events, 2 of which were complicated by the development of acute chest syndrome that resulted in transfer to the intensive care unit. He has missed many school days and may be retained a grade this year. He feels particularly sad about missing the school field trip that occurred during his last hospitalization. He also reports that he is not able to keep up with his friends when participating in physical activities at school. The child's parents are worried that he may be depressed. You as the provider discuss the option of hydroxyurea therapy. His parents ask if hydroxyurea would improve his overall well-being and functioning.
Introduction
Sickle cell disease (SCD), an inherited hemolytic anemia, is associated with multiple acute and chronic complications such as painful vasoocclusive events, cerebral vasculopathy, priapism, and renal or lung disease. These complications are variable and unpredictable and can be associated with significant morbidity and poor quality of life.1 Hematological parameters associated with hydroxyurea (HU) therapy include increases of fetal hemoglobin and mean corpuscular volume and decreases in neutrophil and reticulocyte counts.2 Given as a once-daily oral medication, it is generally well tolerated and is effective in reducing the frequency of painful episodes, acute chest syndrome, and the need for transfusion in adults and children.3–6 Many ongoing studies (www.clinicaltrials.gov) are evaluating the role of HU in preventing and treating SCD-related complications.
Health-related quality of life (HRQL) is defined as the patient's perception of his/her well-being and level of functioning compared with a perceived ideal and as affected by his/her health. Measures of HRQL are multidimensional and include physical, emotional, and social components, along with school/work functioning. Measuring HRQL as an outcome in therapeutic trials has become increasingly common, as noted in ongoing and upcoming studies of SCD.7 Many ongoing studies have proposed to use measures such as the Adult Sickle Cell Quality of Life Measurement Information System (ASCQ-Me) or the Pediatric Quality of Life Inventory (PedsQL) SCD module specifically developed to measure HRQL in patients with SCD.8
To examine the effect of HU on HRQL in SCD, we performed a comprehensive computerized literature search restricted to human studies and the English language using the keywords “sickle cell, ” “quality of life, ” and “HU ” or “hydrea ” or “hydroxycarbamide ” in the first week of June 2012. This strategy yielded a total of 189 citations in PubMed (25 hits), Embase (67 hits), Scopus (65 hits), Web of Science (31 hits), and Psychinfo (I hit). The deletion of duplicate articles (97) left 92 articles. Three studies met the criteria of being research studies of HRQL in patients with SCD receiving HU therapy (Table 1). To check for any ongoing research on HU and HRQL in SCD, we also searched www.clinicaltrials.gov using the same keywords, which identified 9 additional studies in various stages.
Study characteristics and main conclusion of the research studies of HRQL in adults and children with SCD
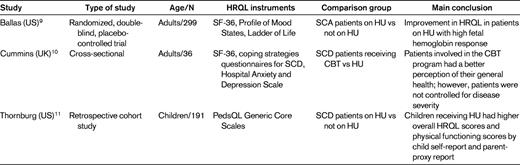
SCA indicates sickle cell anemia; CBT, cognitive behavioral therapy.
Only 1 of the 3 studies was a randomized, double-blind, placebo-controlled trial, the Multicenter Study of Hydroxyurea in Sickle Cell Anemia (MSH) trial, which enrolled 299 adults with moderate to severe disease. The HRQL analysis included 277 patients.9 The Health Status Survey Short Form 36 (SF-36), Profile of Mood States, and Ladder of Life measures were collected at baseline before starting HU or placebo and every 6 months for up to 2 years, during which time patients received randomized treatment. Over 2 years of treatment, improvement in HRQL was limited to those patients receiving HU who had a high hemoglobin F response compared with those with low hemoglobin F response or placebo. The benefits were restricted to certain aspects of HRQL (social function, pain recall, and general health perception).
One of the retrieved articles was a cross-sectional exploratory study in London that examined HRQL in 36 adults with SCD who had received cognitive behavioral therapy (n = 21) or HU (n = 15) as part of their routine clinical care.10 Patients completed the SF-36 and the Coping Strategies Questionnaire, along with other assessments, and comparisons in HRQL were then made between these 2 groups. The investigators concluded that cognitive behavioral therapy appears to improve HRQL in patients with SCD and has the additional advantage of increasing psychological coping ability, and therefore may be a beneficial adjunctive to HU. However, this study was cross-sectional and did not account for time on HU or disease severity, making conclusions regarding the impact of HU on HRQL difficult. In addition, for patients to be on HU in that study, they had to meet strict disease criteria that included frequent pain and hospitalizations, whereas cognitive behavioral therapy was offered as recommended by the psychologist based on self-reported level of pain and impact of SCD on daily living.
The third article retrieved was a pediatric retrospective cohort study comparing the HRQL of children with SCD receiving HU with children not receiving HU. The study used the PedsQL Generic Core Scales parent-proxy report and the child self-report questionnaire, which has been validated for use in children with SCD. It measures physical, social, emotional, and school functioning and higher scores represent better HRQL. After adjusting for age and disease severity, children receiving HU had higher overall HRQL scores (median) (75 vs 69; P = .04) and physical functioning scores (79.7 vs 71.4; P = .01) by child self-report as well as by parent-proxy-report of physical functioning (75 vs 71.9; P = .05). There were no differences in emotional, social, or school functioning.11
Based on our analysis of the literature, we conclude that HU may contribute to improvement in HRQL of children and adults living with SCD. However, this conclusion is based on a very limited number of studies performed in this area. Therefore, we recommend that the decision to initiate HU therapy should be based on clinical indications and not solely on the intent of improving HRQL (grade 2C). Furthermore, evaluating a patient's HRQL at baseline and while receiving HU could help in assessing improvement in HRQL of children and adults with SCD. This review also highlights the current lack of data and the need for further research in this important field.
Disclosures
Conflict-of-interest disclosure: The authors declare no competing financial interests. Off-label drug use: HU is approved for use in SCD in adults but not children.
Correspondence
Deepika S. Darbari, MD, Division of Hematology, Center for Cancer and Blood Disorders, Children's National Medical Center, 111 Michigan Ave NW, Washington, DC 20010; Phone: 202-476-2800; Fax: 202-476-5685; e-mail: ddarbari@cnmc.org.
