
Abstract
Indolent B-cell lymphomas that are supposed to derive from the marginal zone (marginal zone lymphomas [MZLs]) include 3 specific entities: extranodal marginal zone lymphoma (EMZL) or mucosa-associated lymphatic tissue (MALT) lymphoma, splenic MZL (SMZL), and nodal MZL (NMZL). The clinical and molecular characteristics are different for each entity, with some shared phenotypic and genetic features. EMZL is the most common entity, accounting for approximately 70% of all MZLs. These neoplasms can arise at virtually any extranodal site and are commonly associated with chronic antigenic stimulation either as a result of infection (eg, Helicobacter pylori in the stomach) or autoimmune disease (eg, Sjögren syndrome and salivary glands). Several chromosomal translocations were also identified in EMZL, accounting in the aggregate for approximately one-third of all cases. SMZL accounts for approximately 20% of all MZLs. Patients typically present with an enlarged spleen and involvement of abdominal lymph nodes and BM. Approximately 40%-50% of SMZLs are associated with deletions of chromosome 7q. NMZL is the less common entity, representing approximately 10% of all MZLs. Patients with NMZL, by definition, have lymph node–based disease without involvement of the spleen or extranodal sites. The molecular pathogenesis of NMZL is still unknown.
Introduction
Marginal zone lymphomas (MZLs) represent a group of lymphomas that originate from memory B lymphocytes normally present in a distinct micro-anatomic compartment called the “marginal zone” of the secondary lymphoid follicles. MZL develops in spleen and mucosa-associated lymphoid tissues, whereas it is rarely identifiable in lymph nodes.1 According to involved sites and to characteristic molecular findings, the last lymphoma classification2 singles out 3 subtypes of MZLs: extranodal MZL of mucosa-associated lymphoid tissue (MALT) type, splenic MZL (SMZL), and nodal MZL (NMZL). In addition, the recent 2008 World Health Organization (WHO) classification2 introduced a new provisional category of unclassified splenic lymphoma for overlapping entities such as splenic diffuse red pulp lymphoma and hairy cell leukemia-variant.
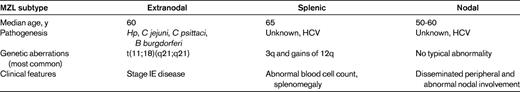
In adults, MZLs account for 5%-17% of all non-Hodgkin lymphomas (NHLs), depending on the series. MALT lymphoma comprises 7%-8% of all B-cell lymphomas3 and it is the third most common NHL. Most cases occur in adults, with a median age of 60 years and a slight female preponderance.3 A geographical variability seems to exist in terms of incidence of gastric MALT lymphomas, with a higher incidence in some areas, for example, northeast Italy.4 Splenic and nodal MZLs represent 20% and 10% of MZLs, respectively, and account for less than 2% of all NHLs.5 The median age of occurrence for SMZL is 65 years6 ; for NMZL, it is and between 50 and 60 years.7 Despite these advances in histologic classification, patients with generalized disease at diagnosis cannot be ascribed easily to a precise diagnostic group.
MALT lymphoma (or extranodal MZL)
MALT lymphoma differs from its splenic and nodal counterparts in that it arises in organs (eg, stomach, lungs, salivary glands, and lachrymal glands) that normally lack lymphoid tissue but have accumulated B cells in response to chronic infections or autoimmune processes.
Pathogenesis
In addition to inducing an initially polyclonal B-cell proliferation, sustained (auto)antigenic stimulation may also trigger inflammatory responses by attracting neutrophils, which release reactive oxygen species. The latter are genotoxic and may cause a wide range of genetic abnormalities. Moreover, prolonged proliferation of B cells induced by chronic inflammation may also increase the risk of DNA damage such as double-strand breaks and translocations due to the intrinsic genetic instability of B cells during somatic hypermutation and class-switch recombination. Remarkably, the genes targeted by most of the abnormalities are involved in the same pathway leading to the activation of NF-κB. The latter is a key transcription factor in the immune response, because it regulates the expression of several survival- and proliferation-related genes in B cells.8 Therefore, its constitutive activation by MALT lymphoma–related genetic abnormalities results in uncontrolled B-cell proliferation and subsequent neoplastic transformation of the B-cell clone.
Long-standing (auto)antigenic stimulation explains how lymphoid infiltrates may appear in extranodal sites that are normally devoid of lymphoid tissue (eg, stomach, lungs, salivary glands, and lachrymal glands). The list of microbial species associated with MALT lymphoproliferations now comprises at least 6 distinct members: Helicobacter pylori (Hp), Helicobacter heilmannii, hepatitis C virus, Campylobacter jejuni, Borrelia burgdorferi, and Chlamydia psittaci, which have been found to be associated with gastric MALT lymphoma, immunoproliferative small intestinal disease (IPSID), cutaneous MALT lymphoma, and orbital MALT lymphoma, respectively9–12 (Table 1). A very high prevalence (up to 90% of cases) of Hp infection was reported in gastric MALT lymphoma.

Of all of the MALT lymphomas, the infectious etiology of gastric MALT lymphoma has been documented the most extensively. There is now compelling evidence that gastric MALT lymphoma is caused by Hp infection. In fact, gastric MALT lymphoma can be induced in vivo (in murine models) by prolonged Hp infection.13 It may be hypothesized that gastric MALT lymphoma arises from Hp-stimulated, autoreactive B cells. Outside of the stomach, the role of (auto)antigens is less clearly defined. However, new criteria were established by recent molecular advances that take into account the host specificity and putative noncultivability of certain microbial organisms.14 Moreover, recent years have witnessed a significant improvement in our understanding of the link between orbital MALT lymphoma and the intracellular bacterium C psittaci. Not only are monocytes/macrophages infiltrating orbital MALT lymphomas carriers of C psittaci (as shown by electron microscopy, protein chain reaction, immunohistochemistry, and fluorescence), but C psittaci is both viable and infectious in the blood and conjunctiva of orbital MALT lymphoma patients.15 Furthermore, it is well established that autoimmune diseases increase the risk of developing nongastric MALT lymphomas. Autoreactive B cells infiltrate the tyroid gland in Hashimoto thyroiditis and the salivary glands in Sjögren syndrome and organize progressively into a MALT-mimicking lymphoproliferation. Patients with Sjögren syndrome have a 44-fold increased risk of developing lymphoma and patients with Hashimoto thyroiditis have a 70-fold increased risk of thyroid lymphoma.16,17
Hp appears to be a fundamental factor for the development of gastric MALT lymphoma. However, as for gastric cancer, because only a minority of people with the infection develop the disease, it is obvious that the lymphoma pathogenesis also depends upon other factors. These factors, largely unknown, may be related to the host, to the environment, or to the virulence of the infecting Hp strain.
Genetic aberrations
MALT lymphomas present with a series of recurrent genomic lesions, including chromosomal translocations and unbalanced aberrations (Table 2).18–22 The t(11;18)(q21;q21) lesion is the most common structural chromosomal abnormality in MALT lymphoma; it is demonstrated in 10%-50% of gastric MALT lymphomas and occurs only rarely in nongastric lymphomas, with the exception of pulmonary MALT lymphomas. The presence of t(11;18)(q21;q21) in MALT lymphomas is correlated with the lack of any further genetic instability or chromosomal imbalances. New data show that the t(11;18)(q21;q21) lesion can be found in both gastric MALT lymphomas and gastric large B-cell lymphomas at approximately equivalent frequencies.23 The chromosomal translocations are mutually exclusive and, unlike 3/3q and 18/18q gains and 6q23 deletions, they show a different anatomical distribution.22

Based on the observations from previous work and on current knowledge of the genetic lesions of MALT lymphoma, a putative model of the multistage development and progression of gastric lymphoma from the background of a chronic gastritis can be proposed. The accumulation of genetic abnormalities is associated with a loss of dependency from antigenic stimulation (with subsequent antibiotic resistance) and with a possible histologic transformation.
Clinical features
MALT lymphomas mostly present as Ann-Arbor stage IE disease (ie, extranodal disease limited to the site of origin), and BM and peripheral lymph node involvement are rather uncommon. The stomach is the most common site of localization, accounting for approximately one-third of cases. Other typical presentation sites include the salivary glands, ocular adnexa, thyroid, lungs, skin, breast, liver, and other gastrointestinal (GI) sites. Advanced disease at diagnosis appears to be more common in MALT lymphomas that arise outside of the GI tract. Up to 25% of patients with gastric lymphoma—but nearly 50% of those with non-GI lymphoma—present with disseminated disease.24 Within the stomach, MALT lymphoma is often multifocal, which may explain the rates of relapses in the gastric stump after surgical excision. Concomitant GI and non-GI involvement can be detected in approximately 10% of cases.25 BM involvement is reported in up to 20% of cases.
The clinical aspects and presenting symptoms of extranodal MZL are generally related to the primary location. Specifically, for gastric MALT lymphomas, the most common presenting symptoms are nonspecific dyspepsia, epigastric pain, nausea, and chronic manifestations of GI bleeding, such as anemia. MALT lymphoma often involves the antrum, but may occur in any part of the stomach. It can appear as intragastric nodularities or enlarged rugal folds. In other cases, it appears as superficial irregularly shaped erosions or shallow ulcers. Overt distant dissemination is not common in cases of gastric MALT lymphomas; commonly involved sites include the BM, small intestine, liver, and spleen.
A special variant of MALT lymphoma is IPSID, which occurs mainly in the Middle East, especially in the Mediterranean area, where the disease is endemic, affecting young adults and predominantly men. IPSID usually manifests with severe, unremitting malabsorption; the lymphoma is characteristically confined to the upper intestine and regional lymph nodes and may rarely spread beyond the abdomen only in advanced stages of the disease, when high-grade transformation occurs.
Treatment and outcome
Approximately 30%-50% of patients with Hp-positive gastric MALT lymphoma show persistent or progressing lymphoma even after eradication of the Hp with antibiotic therapy.26,27 Between complete responders, almost 15% will relapse within 3 years, suggesting that approximately 50% of patients with gastric MALT lymphoma are eventually considered for additional therapies. Patients who present with no evidence of Hp infection are unlikely to respond to antibiotics and should be considered for alternative treatments. The choice should be based on the epidemiology of the infection in the patient's country of residence, taking into account the locally expected antibiotic resistance. The most common approach is a triple therapy: a proton pump inhibitor in association with amoxicillin and clarithromycin. The role of additional chemotherapy after antibiotics was reported in a randomized study comparing chlorambucil versus observation after anti-Hp treatment; in that study, chlorambucil did not increase the progression-free or overall survival rates.28
There is no consensus for the treatment of patients with gastric MALT lymphoma requiring further treatment beyond Hp eradication nor for treatment of patients with nongastric MALT lymphoma.
For patients with early-stage MALT lymphoma of the stomach without evidence of Hp infection, those with persistent lymphoma after antibiotics, and for most nongastric localized presentations, a modest dose of involved-field radiotherapy (25-35 Gy) gives excellent disease control.29,30
In the last decade, the role of surgery in gastric lymphoma was questioned.31,32 Gastric MALT lymphoma is a multifocal disease and adequate gastrectomy needs to be quite extensive, severely impairing quality of life. Residual disease at the margins may still require additional radiation and/or chemotherapy. Table 3 summarizes relatively large series of patients with gastric MALT lymphoma treated with chemotherapy/immunotherapy.33–39 Among nongastric MALT lymphoma, fludarabine has demonstrated some antitumor activity.40
Chemotherapy/immunotherapy in gastric MALT lymphoma
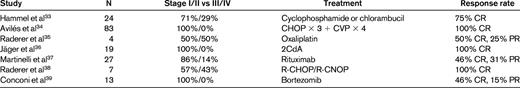
CR indicates complete response; PR, partial response.
The efficacy of the combination of rituximab with chlorambucil was evaluated in a randomized study (comparator was chlorambucil alone) by the International Extranodal Lymphoma Study Group (IELSG) in gastric MALT lymphomas that had failed antibiotics and in nongastric MALT lymphomas. The preliminary report41 showed that the 5-year event-free survival was significantly better for patients treated with chlorambucil plus rituximab.
MALT lymphoma usually has a favorable outcome, with overall survival at 5 years more than 85% in most series. The reported median time to progression is better for GI than for non-GI lymphomas, but with no significant differences in overall survival between the 2 groups. Histologic transformation to large-cell lymphoma has been reported in approximately 10% of the cases, usually as a late event that is independent from dissemination.
Regarding antibiotic treatment in localized, nongastric MALT lymphomas, the finding that C psittaci has a potential pathogenic role in the development of MALT lymphoma of the ocular adnexa and that it was detected in approximately 80% of Italian patients may represent a strong rationale for antibiotic treatment of localized lesions.42 At the same time, the prevalence of C psittaci infection in ocular adnexal lymphoma varies among countries and among different regions within the same country. For example, in the United States, C psittaci was not detected in any case included in 4 North-American series. A prospective phase 2 study was conducted by IELSG and preliminary interesting results show lymphoma regression in more than 60% of patients after doxycycline treatment.43 Lymphoma regression after doxycycline therapy was observed in some lymphomas with no evidence of C psittaci and in cases in which this treatment failed to eradicate C psittaci infection.
The role of high-dose therapy/autologous hematopoietic stem cell transplantation for MZLs is unclear because of a paucity of data (which are for the most part represented by MALT lymphoma). Outcomes of high-dose therapy/autologous hematopoietic stem cell transplantation in patients with disseminated lymphoma are quite similar to those in follicular lymphoma patients.44,45
SMZL
SMZL is a B-cell neoplasm consisting predominantly of small cells and involving the white pulp follicles of the spleen, splenic hilar lymph nodes, BM, and, often, the peripheral blood.
Pathogenesis
The precise pathogenesis of SMZL is unknown. The origin is a marginal zone memory B cell and, in most cases, it is supposed to have a postgerminal origin, as demonstrated by the study of somatic mutations in Ig variable heavy chain region genes46,47 ; however, one-third of cases are nonmutated. Conversely, SMZL exhibits a low frequency of somatic mutations involving some oncogenes (ie, BCL-6, PAX5, PIM1, and RHO-H)48 , suggesting a particular differentiation pathway that may not involve transit through the germinal center.48,49
In SMZL associated with HCV, it was demonstrated that the E2 glycoprotein of HCV could interact with CD81 in the B cells and be responsible for B-cell activation through the BCR, leading to increased proliferation of B cells themselves.50 A decrease in lymphoproliferation after antiviral treatments reinforces the data suggesting a contribution of chronic antigenic stimulation to the pathophysiologic process of HCV-related MZL.51 A special form of SMZL related to HCV has been shown to be correlated with the presence of cryoglobulin.50
Cytogenetic data and molecular abnormalities
Cytogenetic abnormalities are present in 80% of SMZL patients. The most frequent are complete or partial trisomy of 3q (30%-80% of patients) and gains of 12q (15%-20% of patients).52,53 The abnormality considered typical of SMZL, being reported in 40% of the patients, consists of deletion or translocation of chromosome 7q32.52,53 Other cytogenetic alterations involving the chromosomes 8 (9p34, 12q23-24, 18q, and 17p) have been reported53 ; these cytogenetic abnormalities, although not considered typical, may be helpful for the diagnosis of SMZL. SMZL presents a specific transcriptional profile compared with other lymphomas: this peculiar molecular signature includes genes involved in the signaling cascade of the AKT1 pathway,54 but also the BCR signaling pathway, TNF, and NF-κB targets.55
Clinical features
Most SMZL patients look for medical attention due to an abnormal blood cell count, especially due to anemia and/or thrombocytopenia that is more related to splenic sequestration than to BM infiltration and always associated with lymphocytosis. Patients are asymptomatic, but splenomegaly is detectable at clinical examination. In advanced cases, the typical clinical presentation consists of massive splenomegaly frequently associated with small splenic hilar lymph nodes.
SMZL is also associated with autoimmunity: the neoplastic B cells can produce autoantibodies, and a hemolytic autoimmune anemia or autoimmune thrombocytopenia is present in a subset of patients (10%-15%). A relevant percentage of patients (10%-40%) have a serum monoclonal paraprotein (M-component).56
SMZLs with numerous basophilic villous cells in the peripheral blood, formerly denominated as splenic lymphoma with villous lymphocytes, are characterized by a peculiar histology with atrophic white pulp and a monomorphic diffuse infiltration of a congested red pulp that is reminiscent of the hairy cell leukemia variant. A few differences are found in the clinical presentation, including significantly older age and the absence of immune disorders.
Treatment and outcome
The median overall survival for SMZL is 5-10 years, but in cases of aggressive disease (25%-30% of cases), the median survival is less than 4 years.57 The Italian Foundation of Lymphomas (FIL) has developed a prognostic model based on the tracking of 3 factors (hemoglobin level less than 12 g/dL, lactate dehydrogenase level greater than normal, and albumin level less than 3.5 g/dL) in more than 300 patients, setting up a prognostic index.58 This index allows patients to be separated into 3 subsets, each with a different 5-year survival rate: 88% in the low-risk group (no risk factors), 73% in the intermediate-risk group (1 risk factor), and 50% in the high-risk group (more than 1 risk factor). So far, this index has not yet been shown to have any therapeutic implications. Histologic transformation to large B-cell lymphoma is reported in 10%-20% of patients.
Treatment is required only in symptomatic SMZL patients with large splenomegaly that is associated or not with cytopenia due to hypersplenism. Asymptomatic patients may be followed for several years with clinical examination and blood counts. The absence of treatment does not influence the course of disease. When a treatment is indicated due to the occurrence of clinical symptoms, the recommended frontline therapy is splenectomy.59,60 Patients achieve only a partial response, with a persisting BM and blood lymphocytosis, but with a correction of anemia, thrombocytopenia, and neutropenia. This status can be maintained for years, with a median time to next treatment of 8 years.61 Chemotherapy may be proposed to patients with contraindications to surgery, elderly patients, or those who have progression after surgery. Regimens are based on alkylating agents (eg, chlorambucil or cyclophosphamide), fludarabine, or rituximab both as a single agents and combined with chemotherapy.62–65 Recently, bendamustine was shown to have activity in SMZLs.66 On the basis of these preliminary data, in the next few months, an IELSG phase 2 prospective study will begin with the aim of assessing the safety and the efficacy of the combination of rituximab and bendamustine in symptomatic patients with SMZL not eligible for or not willing to undergo splenectomy.
Nodal MZL
The present WHO lymphoma classification2 considers NMZL as a distinct clinicopathologic subtype within the wide spectrum of marginal zone–derived lymphomas. “Conditio sine qua non” for this diagnosis is primary lymph node localization in the absence of previous or concurrent involvement of any extranodal site, with the exception of BM.
Cytogenetic and molecular findings
No specific diagnostic hallmarks of NMZL have been reported. NMZL may show different patterns of lymph node infiltration, including marginal zone–like/perifollicular, nodular, and diffuse ones.67 No typical cytogenetic abnormality of NMZL has been reported. Among the cytogenetic alterations reported are +3, +7, +12, +18, and structural rearrangements of chromosome 1 with break points in 1q21 or in 1p34.68,69 Gain of several regions of chromosome 3 seems to constitute a common marker for NMZL, as reported in a chromosome-based study on NMZL70 ; however, these patients also had extranodal disease. These chromosome 3 abnormalities occur in 20%-25% of NMZL patients. This type of lymphoma does not display SMZL-related 7q losses.54 The majority of NMZL patients (≥ 75%) have somatic mutations of IGHV genes and a biased use of IGHV4-34.49 It has been demonstrated that both HCV-positive and HCV-negative cases of NMZL harbor somatic mutations in the Ig variable heavy chain region genes, but with usage of different segments between the 2 groups, indicating different antigenic stimulation in lymphoma B-cell precursor selection.71
Clinical features
Given the recent identification of NMZL, there are few studies detailing clinical and outcome data. Only 9 clinical series are available.72–79 The majority of these patients presents with disseminated peripheral and abdominal nodal involvement. BM involvement occurs in less than half of the patients and peripheral blood involvement is quite rare. Performance status is generally good and B-cell symptoms are reported, with percentages ranging from 10%-40%. A serum M-component is detected in only 10% of patients. HCV infection is reported in association with NMZL.80
Treatment and outcome
The average 5-year overall survival of NMZL is approximately 60%-70%, with an estimated 5-year event-free survival of approximately 30%. Relapse at extranodal sites is rare. Biological characteristics of the tumor cells found to be significantly associated with survival are decreases in survivin and active caspase 3 and overexpression of cyclin E.49 Both the International Prognostic Index (IPI) and the Follicular Lymphoma International Prognostic Index (FLIPI) discriminate patients with high and low risk.
No treatment consensus guidelines have been developed for NMZLs, but patients may be managed according to guidelines established for follicular lymphomas. In limited-stage disease, surgery and radiotherapy seem appropriate; in advanced-stage disease, immunochemotherapy is a powerful option. Among the new drugs, bortezomib has demonstrated activity in NMZL.81 Veltuzumab, a humanized anti-CD20 Ab, was used in a few cases of NMZL.82 In relapsed young patients, high-dose therapy and autologous transplantation could be considered.83 In patients with NMZL and HCV-related chronic hepatitis not needing immediately chemotherapy for lymphoma, an antiviral treatment with pegylated IFN and ribavirin is recommended.84
Conclusions
In the past 2 decades, extraordinary progress has been made in our understanding of the etiology and critical cellular and molecular pathological events of MZLs (Table 4). However, there is a lack of large databases or clinical data on patients with these lymphomas because of their rarity. The establishment of national and international study groups has improved our clinical understanding of MZLs, but there is a need for such groups to better decide treatment strategies.
Disclosures
Conflict-of-interest disclosure: The author has received research funding from Spectrum; has consulted for Celgene and Millennium Takeda; and has been affiliated with the speakers' bureaus for Millennium, Takeda, Mundi Pharma, Roche, Pfizer, and Janssen. Off-label drug use: bortezomib, lenalidomide, BTK inhibitors, CAL-101 yttrium-90, ibritumomab, and tiuxetan.
Correspondence
Pier Luigi Zinzani, Istituto di Ematologia “Seràgnoli” Università di Bologna, Via Massarenti 9, 40138 Bologna, Italy; Phone: +39-0516363680; Fax: +39-0516364037; e-mail: pierluigi.zinzani@unibo.it.
