
Abstract
Acute myeloid leukemia (AML) with t(8;21) or inv(16) is commonly referred to as core-binding factor AML (CBF-AML). The incorporation of high-dose cytarabine for postremission therapy has substantially improved the outcome of CBF-AML patients, especially when administered in the setting of repetitive cycles. For many years, high-dose cytarabine was the standard treatment in CBF-AML resulting in favorable long-term outcome in approximately half of the patients. Therefore, CBF-AML patients are generally considered to be a favorable AML group. However, a substantial proportion of patients cannot be cured by the current treatment. Additional genetic alterations discovered in CBF-AML help in our understanding of the process of leukemogenesis and some of them may refine the risk assessment in CBF-AML and, importantly, also serve as targets for novel therapeutic approaches. We discuss the clinical and genetic heterogeneity of CBF-AML, with a particular focus on the role of KIT mutations as a prognosticator, and also discuss recent efforts to target the KIT kinase in the context of existing therapeutic regimens.
Introduction
Acute myeloid leukemia (AML) with t(8;21)(q22;q22) and with the pericentric inversion of chromosome 16 [inv(16)(p13.1q22)] or the less frequent balanced translocation t(16;16)(p13.1;q22) [hereafter collectively referred to as inv(16)], are recognized by the World Health Organization classification as unique entities within the category “AML with recurrent genetic abnormalities.”1 Upon detection of these clonal genetic abnormalities, the diagnosis of AML can be made regardless of the proportion of BM blasts.1 Among adults with de novo AML, t(8;21) and inv(16) are found in 7% and in 5% to 8% of the patients, respectively.2,3 The frequency of t(8;21) and inv(16) decreases in older patients, and only ∼ 7% of AML patients above the age of 60 years harbor one of both chromosome aberrations.4 Both the t(8;21) and inv(16) aberration result in formation of novel chimeric fusions involving genes of the core-binding factor (CBF) complex, a master regulator of definitive hematopoiesis providing the common designation “CBF-AML.” Compared with other cytogenetic AML groups, patients with CBF-AML are considered a favorable AML risk group based on their high remission rate and survival probabilities. However, because approximately one-half of patients with CBF-AML are still not cured, there is a need for markers to identify patients unlikely to respond to current treatment and to develop novel therapeutic approaches based on a better understanding of pathophysiology of the disease.
Treatment and outcomes
Chemotherapy
After anthracycline- and cytarabine-based induction chemotherapy, ∼ 90% of both t(8;21) and inv(16) AML patients achieve a complete remission (CR).5-7 A pioneering study from the Cancer and Leukemia Group B (CALGB) has reported that postremission therapy with high dose of cytarabine (HiDAC; 3 g/m2 bid on days 1, 3, and 5) resulted in a clear survival advantage in CBF-AML compared with intermediate-dose cytarabine (IDAC) or lower doses (400 or 100 mg/m2, respectively, as a continuous infusion on days 1 to 5).8 In a subsequent CALGB study, the administration of repetitive cycles of HiDAC compared with only one cycle was shown to reduce the risk of relapse both in t(8;21) and inv(16) AML, but was not associated with a significant prolongation in overall survival (OS).6 A more recent Japanese study provides further evidence for the benefit of HiDAC in postremission treatment of CBF-AML compared with low-dose cytarabine.9 In this study, postremission treatment with 3 courses of HiDAC (2 g/m2 bid on days 1 to 5) was compared with 4 courses of polychemotherapy including cytarabine at a dose of 200 mg/m2 (as a continuous infusion on days 1 to 5)9 ; treatment with HiDAC was associated with a superior disease-free survival (DFS; at 5 years: 57% vs 39%), but this did not translate into OS improvement. The implementation of repetitive cycles of HiDAC for postremission treatment results in ∼ 50% of CBF-AML patients achieving long-term survival6,7 and is currently used by several cooperative groups as standard primary treatment in first CR (CR1) for this patient subset. However, several cycles of HiDAC chemotherapy frequently causes considerable toxicities, particularly in older patients, and intensively dosed cytotoxic chemotherapy can thus often be administered to only a minor proportion of patients. In addition, there is still the open question of how much cytarabine is enough to effectively treat CBF-AML without jeopardizing the outcome benefit. There is some evidence that less cytarabine is sufficient for an effective treatment of CBF-AML without decreasing the outcome benefit. A recent study by the HOVON/SAKK group (Dutch Belgian Cooperative Trial Group for Hemato-Oncology/Swiss Group for Clinical Cancer Research) showed similar outcome results for CBF-AML patients treated by multiagent chemotherapy incorporating cytarabine at a cumulative dose of 13.4 g/m2 (IDAC) and 26 g/m2 (HiDAC).5 It should be noted that cytarabine in this study was administered in 2 treatment courses. Analyses of the CBF-AML subset in this study have shown similar event-free survival (EFS) and OS for patients treated with IDAC and HiDAC (EFS at 5 years: 58% vs 47%; OS at 5 years: 64% vs 67%). Based on the data we have so far, the optimal dose of cytarabine and number of chemotherapy courses is not commonly defined for the treatment of CBF-ABL.
A study from the MD Anderson Cancer Center on 114 patients with CBF-AML reported improved EFS in patients treated with fludarabine and cytarabine with (FLAG) or without (FA) G-CSF compared with chemotherapy containing idarubicin and cytarabine with (IAG) or without (IA) G-CSF. Among the 107 patients achieving a CR, relapses occurred in 32% of the patients treated with FLAG, in 33% treated with FA, and in 52% treated with IA/IAG.10
Gemtuzumab ozogamicin
The immunoconjugate gemtuzumab ozogamicin (GO) combines an antibody directed against the CD33 antigen with calicheamicin, a DNA-damaging toxin.11 Upon binding to CD33, the CD33-GO complexes are rapidly internalized, providing a convenient drug-delivery mechanism into the cell. This explains why cells expressing higher levels of CD33 are more susceptible to GO.11 However, because patients with CD33-negative AML respond to treatment with GO, a CD33-independent endocytotic transport of the drug into the cells has been suggested.11 GO was approved by the Food and Drug Administration in the year 2000 for the treatment of relapsed AML in patients older than 60 years who were unfit for intensive therapy, but due to safety concerns raised by an interim analysis of a following randomized trial,12 the drug was withdrawn from the market in 2010. The Medical Research Council (MRC) AML15 trial, in which younger AML patients (< 60 years) were randomized to receive one dose of GO (3 mg/m2) in addition to induction and/or to the third course of chemotherapy, did not show any increased toxicity or higher early death rate in patients assigned to receive GO.13 Overall, the addition of GO did not affect outcome in AML. However, in subset analyses, a significant survival benefit was observed in CBF-AML patients who did receive GO compared with the CBF-AML group who did not receive GO (OS at 5 years 79% vs 51%).13 The reasons why the beneficial effect of GO on OS was confined to this AML subgroup are unclear, but the presence and degree of CD33 blast positivity were revealed not to be predictive factors in this study.13 Further studies are needed to confirm the subgroup-dependent beneficial impact of GO in CBF-AML and to elucidate its mechanisms of action. To date, GO has not been reapproved for the treatment of AML and is currently only available within clinical trials. However, the accumulated data on the efficacy of GO in newly diagnosed AML, particularly in favorable-risk AML and in acute promyelocytic leukemia, and also on its acceptable toxicity profile, provide a basis for its reapproval.14 In addition, alternative anti-CD33 antibodies such as lintuzumab (NCT00014495, NCT00038051, NCT00528333)15-17 and BI 836858 (NCT01690624) have been or are being explored in clinical trials.
Allogeneic and autologous stem cell transplantation
Allogeneic stem cell transplantation (SCT) is generally not administered in patients with CBF-AML in CR1, a concept that is supported by different studies. A meta-analysis of several prospective trials evaluating the impact of allogeneic SCT for AML in CR1 did not show a benefit of allogeneic SCT on relapse-free survival (RFS) and OS for favorable-risk AML (n = 547) compared with nonallogeneic SCT therapies including postremission chemotherapy, autologous SCT, or both.18 This is consistent with the results of a recent MRC study, where no survival benefit in CBF-AML patients who underwent allogeneic SCT in CR1 was observed compared with patients not receiving HCT.19 Another retrospective analysis of younger (<60 years) AML patients with t(8;21) compared the outcomes of 118 patients who received allogeneic SCT from a HLA-identical sibling with 132 patients treated with cytarabine-based chemotherapy within 8 German AML trials.20 After allogeneic SCT, the relative risk (ReR) of relapse was significantly lower (P = .014; ReR = 0.5), but the ReR of treatment-related mortality was significantly higher (P < .001; ReR = 6.8) compared with chemotherapy.20 No benefit regarding RFS and OS was found for allogeneic SCT in the entire study cohort.20 However, OS in this study differed between patients treated with allogeneic SCT and chemotherapy when secondary chromosome aberrations were considered. Although the OS was similar for both types of postremission treatment in t(8;21) patients with the loss of a sex chromosome (LOS), patients without LOS treated with allogeneic SCT had a significantly higher risk of overall mortality (P = .002; ReR = 3.5) than patients treated with chemotherapy.20 Two other studies compared the results of allogeneic versus autologous SCT in t(8;21) and inv(16) AML. A study by Gorin et al reported a significantly higher relapse incidence after autologous SCT compared with allogeneic SCT in t(8;21) (P = .03; 28% vs 15%) but not in inv(16) AML.21 In this study, the treatment-related mortality after allogeneic SCT was significantly higher in both t(8;21) (P = .003; 24% vs 6%) and in inv(16) (P = .003; 14% vs 2%) AML, but the type of transplantation did not affect leukemia-free survival (LFS).21 A Japanese study also showed comparable results for OS after allogeneic and autologous SCT in CR1 for both t(8;21) and inv(16) AML.22
Salvage treatment in relapsed CBF-AML
Overall, 60% to 85% of patients with CBF-AML experiencing a relapse achieve a CR2 after salvage therapy.7,19,23 Among relapsed CBF-AML patients, the CR2 rate is higher (86%-97% vs 33%-78%) and the long-term outcome after a second intensive consolidation more favorable in inv(16) AML.6,7,23,24 Relapsing CBF-AML patients who achieve a CR2 frequently undergo allogeneic SCT. However, a recent retrospective MRC study in CBF-AML patients achieving a CR2 failed to demonstrate a survival benefit for allogeneic SCT compared with non-allografted patients.19 A Japanese study analyzing the outcome of CBF-AML patients undergoing allogeneic SCT in CR2 or CR3 reported a better OS in inv(16) than in t(8;21) AML patients (P = .008; 3-year OS 86% vs 45%), and in trend also a better DFS (P = .053; DFS at 3 years 71% vs 43%).22 These results are consistent with the data reported by previous studies, which suggested a higher sensitivity of inv(16) AML to salvage treatment.6,7 The Japanese study also reported that, among patients not being in CR at the time of transplantation, those with inv(16) AML had a significantly better 3-year DFS (P = .02; 75% vs 18%) and OS (P = .03; 70% vs 18%) compared with patients with t(8;21) AML.22
CBF-AML in older patients
In a French study on 147 patients with t(8;21) or inv(16) age 60 years or older, almost 90% achieved a CR after 1 to 2 courses of induction therapy.25 This high CR rate is comparable to what is seen in younger CBF-AML patients, indicating that chemosensitivity is retained in older CBF-AML. However, the LFS at 5 years in the French study was only 26%.25 The group of patients who received intensive postremission therapy, incorporating IDAC to HiDAC, appeared to have a superior LFS compared with those who received maintenance therapy only (P = .08; median LFS 26 vs 14 months).25 This was mainly due to a better LFS in patients with t(8;21) AML but not in AML patients with inv(16).25
Genetic basis of CBF-AML
The CBF is a heterodimeric transcription factor complex composed of a DNA-binding alpha subunit and a beta subunit, which does not bind to DNA, but enhances the DNA affinity of the CBF complex. The CBF alpha subunit is encoded by 1 of 3 homologous genes of the RUNX (Runt-related transcription factor) family, whereas the common CBF beta subunit is encoded by the CBFB (PEBP2B) gene.26 Murine knockout embryos with homozygous loss of either Runx1 or Cbfb allele fail to develop definitive hematopoiesis and die in utero, indicating that both CBF subunits are critical for normal hematopoietic development.27-30 At the molecular level, both t(8;21) and inv(16) result in the formation of novel chimeric fusions involving genes of the CBF complex; the t(8;21) aberration results in the fusion of RUNX1 on chromosome 21q22 with RUNX1T1 (ETO; MTG8) on chromosome 8q22 and the inv(16) mutation results in the CBFB gene on chromosome 16q22 being fused to MYH11 on chromosome 16p13.1.31-33 The CBF fusion proteins are considered to affect the normal CBF function in a dominant-negative manner. This hypothesis is supported by studies on mouse knock-in embryos heterozygous for Runx1-RUNX1T1 or Cbfb-MYH11, which develop a similar phenotype to Runx1 und Cbfb knockouts without definitive hematopoiesis and embryonic death.34,35 Therefore, the formation of CBF fusions is believed to interfere with normal hematopoietic differentiation and to provide a condition predisposing to leukemia.
Cooperating alterations
Full-length RUNX1-RUNX1T1 and CBFB-MYH11 fusions are considered as preleukemic conditions in CBF leukemogenesis, but they alone are not sufficient to induce leukemic transformation. The acquisition of additional genetic hits is necessary for the development of a leukemic phenotype.36,37 Some secondary alterations cooperating with CBF fusion proteins in the process of leukemogenesis are mutations in genes encoding protein effectors controlling cell proliferation and/or conferring survival advantage to the malignant cells. Genes encoding tyrosine kinases, namely KIT (v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog) and FLT3 (FMS-like tyrosine kinase), as well as N- and K-RAS guanosine triphosphatases, that is, NRAS (neuroblastoma rat sarcoma viral oncogene homolog) and KRAS (Kirsten rat sarcoma viral oncogene homolog), have been identified as frequent secondary mutations in CBF-AML. Indeed, almost 90% of AML with t(8;21)38 (Figure 1A) and more than 90% of AML with inv(16)39 (Figure 1B) harbor additional secondary chromosome aberrations and/or mutations affecting KIT, FLT3, NRAS, and KRAS.
![Figure 1. Pie charts illustrating the genetic heterogeneity and coexistence of distinct secondary genetic abnormalities in AML with t(8;21) and inv(16). The charts are based on patients with complete cytogenetic data and complete mutation status on KIT, FLT3, NRAS, and KRAS. (A) Genetic alterations in 141 AML patients with t(8;21) treated on AMLSG trials. Among the secondary chromosome aberrations, loss of a sex chromosome (−Y or −X), deletions of the long arm of chromosome 9 [del(9q)], and trisomy 8 (+8) are indicated; all other secondary chromosome aberrations constitute one group abbreviated in the chart as “o.c.a.” Due to the rounding error, all values do not add up to exactly 100%. (B) Genetic alterations in 166 AML patients with inv(16) treated on AMLSG trials. Among the secondary chromosome aberrations, trisomy 22 (+22) and trisomy 8 (+8) are indicated; all other secondary chromosome aberrations constitute one group abbreviated in the chart as o.c.a. Due to the rounding error, all values do not add up to exactly 100%. (Used with permission from Paschka et al.32 Copyright American Society of Hematology.)](/view-large/figure/6508172/bep0011302730001.jpeg)
Pie charts illustrating the genetic heterogeneity and coexistence of distinct secondary genetic abnormalities in AML with t(8;21) and inv(16). The charts are based on patients with complete cytogenetic data and complete mutation status on KIT, FLT3, NRAS, and KRAS. (A) Genetic alterations in 141 AML patients with t(8;21) treated on AMLSG trials. Among the secondary chromosome aberrations, loss of a sex chromosome (−Y or −X), deletions of the long arm of chromosome 9 [del(9q)], and trisomy 8 (+8) are indicated; all other secondary chromosome aberrations constitute one group abbreviated in the chart as “o.c.a.” Due to the rounding error, all values do not add up to exactly 100%. (B) Genetic alterations in 166 AML patients with inv(16) treated on AMLSG trials. Among the secondary chromosome aberrations, trisomy 22 (+22) and trisomy 8 (+8) are indicated; all other secondary chromosome aberrations constitute one group abbreviated in the chart as o.c.a. Due to the rounding error, all values do not add up to exactly 100%. (Used with permission from Paschka et al.32 Copyright American Society of Hematology.)
Pie charts illustrating the genetic heterogeneity and coexistence of distinct secondary genetic abnormalities in AML with t(8;21) and inv(16). The charts are based on patients with complete cytogenetic data and complete mutation status on KIT, FLT3, NRAS, and KRAS. (A) Genetic alterations in 141 AML patients with t(8;21) treated on AMLSG trials. Among the secondary chromosome aberrations, loss of a sex chromosome (−Y or −X), deletions of the long arm of chromosome 9 [del(9q)], and trisomy 8 (+8) are indicated; all other secondary chromosome aberrations constitute one group abbreviated in the chart as “o.c.a.” Due to the rounding error, all values do not add up to exactly 100%. (B) Genetic alterations in 166 AML patients with inv(16) treated on AMLSG trials. Among the secondary chromosome aberrations, trisomy 22 (+22) and trisomy 8 (+8) are indicated; all other secondary chromosome aberrations constitute one group abbreviated in the chart as o.c.a. Due to the rounding error, all values do not add up to exactly 100%. (Used with permission from Paschka et al.32 Copyright American Society of Hematology.)
KIT
High KIT expression is observed in hematopoietic stem cells.40 KIT, also designated as CD117, is commonly used as a phenotypic marker for hematopoietic stem cells. The KIT protein is a member of type III tyrosine kinases (RTK), which share a common protein structure consisting of 5 immunoglobulin-like domains in the extracellular part, a transmembrane domain, an intracellularly located juxtamembrane (JM), and a split kinase domain.40 Under physiological conditions, the monomeric KIT receptor dimerizes after the binding of its specific ligand, the SCF, and then KIT becomes autophosphorylated at key tyrosine sites and activates downstream signaling pathways including the Ras/ERK, PI3K, Src kinases, and JAK/STAT pathways.40 Both the KIT receptor and SCF are essential for the maintenance of normal hematopoiesis.40
Ligand-independent constitutive KIT activation can be caused by gain-of-function mutations. Such mutations have been detected in various malignancies, including mastocytosis, gastrointestinal stromal tumors, a distinct type of melanoma, germ cell tumors, and AML.40-42 Although in all AML, KIT mutations represent an infrequent molecular alteration,43-45 they can be detected in approximately one-third of patients with CBF-AML (Table 1), representing the most frequently mutated RTK in CBF-AML. The main mutational clusters in CBF-AML include KIT exon 17, which encodes the activation loop (A-loop), and exon 8, which encodes a region in the extracellular part of the receptor. Mutations involving transmembrane domain and JM domains have been only rarely reported in CBF-AML.39,46-49 In vitro and mice studies support that mutant KIT is a sufficient cooperating second event in the development of CBF leukemias. In a cytokine-dependent myeloid cell line (32D), the overexpression of common KIT A-loop and JM mutants induced factor-independent growth and the retroviral infection of NIH3T3 fibroblast cell line with these KIT mutants caused ligand-independent colony formation.50 Recent mice studies provided further evidence for mutant Kit as a sufficient cooperative event in CBF leukemogenesis.50-52 In addition, one study showed that the KitD814V A-loop mutant (corresponding to human KITD816V) and a distinct KIT exon 8 mutant differ with respect to their transforming abilities.52 In that study, the coexpression of Runx1-Run1xt1 and KitD814V resulted in lethal hematopoietic malignancies of short-term latency (2-4 months) in all cases including AML (45%), myeloproliferative neoplasia (35%), and pre-B-acute lymphoblastic leukemia (20%), whereas only half of the mice that coexpressed Runx1-Runx1t1 and the Kit exon 8 mutant developed AML with a latency of 4-5 months within the observation time of 1 year; AML was the only malignant hematological phenotype noticed in these animals.52
Retrospective studies assessing the prognostic relevance of KIT mutations in t(8;21) and inv(16) AML
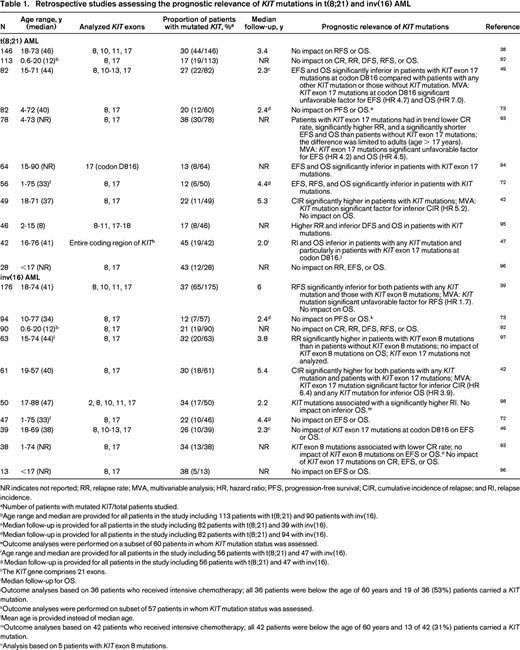
NR indicates not reported; RR, relapse rate; MVA, multivariable analysis; HR, hazard ratio; PFS, progression-free survival; CIR, cumulative incidence of relapse; and RI, relapse incidence.
aNumber of patients with mutated KIT/total patients studied.
bAge range and median are provided for all patients in the study including 113 patients with t(8;21) and 90 patients with inv(16).
cMedian follow-up is provided for all patients in the study including 82 patients with t(8;21) and 39 with inv(16).
dMedian follow-up is provided for all patients in the study including 82 patients with t(8;21) and 94 with inv(16).
eOutcome analyses were performed on a subset of 60 patients in whom KIT mutation status was assessed.
fAge range and median are provided for all patients in the study including 56 patients with t(8;21) and 47 with inv(16).
g Median follow-up is provided for all patients in the study including 56 patients with t(8;21) and 47 with inv(16).
hThe KIT gene comprises 21 exons.
iMedian follow-up for OS.
jOutcome analyses based on 36 patients who received intensive chemotherapy; all 36 patients were below the age of 60 years and 19 of 36 (53%) patients carried a KIT mutation.
kOutcome analyses were performed on subset of 57 patients in whom KIT mutation status was assessed.
lMean age is provided instead of median age.
mOutcome analyses based on 42 patients who received intensive chemotherapy; all 42 patients were below the age of 60 years and 13 of 42 (31%) patients carried a KIT mutation.
nAnalysis based on 5 patients with KIT exon 8 mutations.
Retrospective studies have assessed the impact of KIT mutations as a prognostic marker in t(8;21) and inv(16) AML (Table 1). Although in several but not all studies in t(8;21) AML, KIT mutations, in particular those affecting the A-loop, have been associated with unfavorable outcome, the prognostic impact of KIT mutations in inv(16) AML is less clear (Table 1). In inv(16) AML, the breakpoint variability in the MYH11 gene results at least in 10 different fusion variants, with type A fusion being found in ∼90% of the cases.53 One recent study in adult patients with inv(16) AML reported for the first time that KIT mutations in exons 8 and 17 do not occur in patients with non-type A CBFB-MYH11 fusions.54 In that study, patients with KIT mutations had significantly inferior EFS and OS compared with patients with type A CBFB-fusion and wild-type KIT and patients with non-type-A CBFB fusions.54 In contrast to adult CBF-AML, most pediatric studies did not show prognostic relevance of mutated KIT (Table 1). Although the current data do not yet support the use of KIT mutational status in clinical practice to guide clinical decision making regarding therapeutic interventions, testing for KIT mutations as a prognostic marker has already been implemented into the National Comprehensive Cancer Network (NCCN) guidelines.55 Indeed, according to the current NCCN guidelines, AML with t(8;21) or inv(16) and mutated KIT are considered as intermediate-risk AML, not favorable-risk AML.55 In contrast, within the international European LeukemiaNet (ELN) recommendations, the assessment of KIT mutational status is currently not recommended as part of the initial routine diagnostic workup and this has thus far not affected patient management.56
Global gene expression studies found KIT to be highly expressed in CBF-AML independent of its mutation status57,58 (gene expression data from these studies are available at www.oncomine.org). CBF-AML cases with mutated KIT show even higher KIT expression than those without KIT mutations.59 Therefore, in CBF-AML, there is a rationale to use tyrosine kinase inhibitors (TKIs) to target both the overexpressed and mutated KIT. In vitro studies support the concept that exposure to TKIs inhibits growth of cells expressing wild-type KIT or various KIT mutants.60-66 The rationale to implement KIT inhibitors into treatment concepts for CBF-AML is further supported by a recent murine study in which leukemic cells coexpressing RUNX1-RUNX1T1 and the A-Loop KITN822K mutant were injected into sublethally irradiated mice and the animals were subsequently treated with cytarabine and/or the TKI dasatinib.50 The combination of cytarabine with dasatinib significantly prolonged the survival of the animals compared with treatment with either as single drugs.50 It has been anecdotally reported that the administration of the TKI dasatinib as single agent in a patient with t(8;21) AML and a KITN822K mutation detected at diagnosis, who relapsed after several lines of treatment, resulted in blast clearance, induced differentiation, and stabilized the course of the disease for more than 3 months without any additional chemotherapy.67
In the meantime, trials combining conventional chemotherapy with the therapeutic principle of targeting KIT are ongoing in patients with CBF-AML (NCT01238211, NCT00850382, NCT00651261, NCT01830361). However, when designing treatment trials implementing KIT inhibitors, it needs to be considered that a distinct compound might not be efficient against a particular KIT mutant (Table 2).
In vitro activity of selected compounds against KIT wild-type and distinct KIT mutants
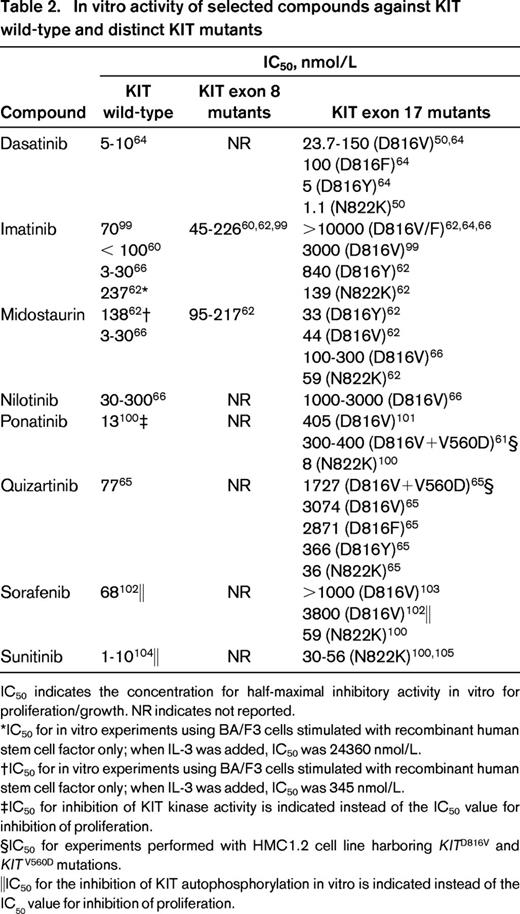
IC50 indicates the concentration for half-maximal inhibitory activity in vitro for proliferation/growth. NR indicates not reported.
*IC50 for in vitro experiments using BA/F3 cells stimulated with recombinant human stem cell factor only; when IL-3 was added, IC50 was 24360 nmol/L.
†IC50 for in vitro experiments using BA/F3 cells stimulated with recombinant human stem cell factor only; when IL-3 was added, IC50 was 345 nmol/L.
‡IC50 for inhibition of KIT kinase activity is indicated instead of the IC50 value for inhibition of proliferation.
§IC50 for experiments performed with HMC1.2 cell line harboring KITD816V and KITV560D mutations.
‖IC50 for the inhibition of KIT autophosphorylation in vitro is indicated instead of the IC50 value for inhibition of proliferation.
FLT3
The FLT3 gene also encodes a type III RTK. A combined analysis of 21 CBF-AML studies reported that activating internal tandem duplications within the JM domain can be found in 7.1% of t(8;21) patients and in 4.9% of inv(16) patients.68 In this combined analysis, activating mutations involving the FLT3 tyrosine kinase domain (TKD) were reported in 3.6% of t(8;21) and in 13.3% of inv(16) patients.68 The results from this study fit very well with our data obtained from large cohorts of patients with t(8;21) and inv(16) AML that were treated within clinical trials of the German AML Study Group (AMLSG).38,39 Recently, additional missense mutations affecting the asparagine 676 in the first part of the kinase domain were identified in 6% (4/69) of patients with inv(16) AML.69 These mutations cannot be detected by routinely performed FLT3 assays, which typically cover the mutational hot spot within the second part of the tyrosine kinase domain (codons D835 and I836).
Mouse transplantation studies have demonstrated that the cotransduction of FLT3-internal tandem duplications with RUNX1-RUNX1T170 and CBFB-MYH1171 promotes leukemia, indicating the cooperative nature of these FLT3 alterations.
The prognostic relevance of FLT3 mutations in CBF-AML is not very well established. In our recent large study on 176 patients with inv(16) AML, we reported that FLT3 mutations as one group are associated with inferior OS in inv(16) AML.39 This adverse effect appeared to be mainly conferred by FLT3-TKD mutations.39 An adverse impact of FLT3 mutations on outcome in inv(16) AML has been suggested previously by 2 smaller studies,72,73 but one MRC study reported that FLT3-TKD mutations confer favorable prognosis in this cytogenetic AML subgroup.74 Although the prognostic relevance of FLT3 mutations in CBF-AML is still controversial, the activated FLT3 kinase represents a further therapeutic target for TKIs (NCT00651261).
RAS
Mutations involving NRAS and, less often, KRAS represent another frequent molecular alteration in CBF-AML. RAS was found mutated in 10% to 20% of AML with t(8;21)38,72,73 and in 35% to 50% of AML with inv(16).39,72,73,75 There is recent evidence from in vitro experiments for the cooperation of oncogeneic NRAS (NRASG12D) with RUNX1-RUNX1T1 in a stepwise leukemic transformation.76 Thus far, RAS mutations have not been shown to be a prognostic marker in CBF-AML.38,39,72,73,77
Casitas B-cell lymphoma
Casitas B-cell lymphoma (CBL) protein is an E3-ligase involved in the degradation of a variety of activated tyrosine kinases. CBL splice sites mutations at exons 8 and 9 leading to variants lacking these exons were found in 3% of t(8;21) and 5% to 16% of inv(16) AML cases.75,78 The mutated CBL might represent an alternative mechanism of tyrosine kinase activation by preventing an appropriate degradation of activated tyrosine kinases such as KIT and FLT3.
Beyond gene mutations
Secondary chromosome aberrations can be detected in more than 60% of t(8;21) and in ∼ 35% to 40% of inv(16) AML cases. The most frequent secondary chromosome aberration in t(8;21) AML is LOS, followed by deletions of the long arm of chromosome 9 [del(9q)] and trisomy 8, whereas the most frequent secondary chromosome aberration in inv(16) AML is trisomy 22, followed by trisomies of chromosome 8 and 21, respectively.41 Thus far, the molecular mechanisms caused by these chromosome aberrations and their contribution to the development of CBF leukemias remain unexplained. However, some candidate genes were identified that could be involved in the process of leukemogenesis. In a recent study from our group, 7q deletions were found to be a relatively frequent alteration in CBF-AML, occurring in ∼ 10% of the cases.79 However, one-half of 7q deletions in this study were only detectable by high-resolution genetic profiling.79 Deletions at chromosome 7q involving the region 7q36.1 were found in almost 8% of the 300 CBF-AML cases analyzed; this chromosome region contains, among others, the MLL3 gene, a putative tumor suppressor.79 Another study using high-resolution genetic profiling in inv(16) AML identified variable submicroscopic deletions at chromosome 17q11 in 16% (6/37) of the cases.75 The only gene in the minimal deleted region was the tumor suppressor gene NF1 (tumor-suppressor gene neurofibromatosis-1).75 The relatively high incidence of NF1 deletions in inv(16) AML found in that study still needs to be confirmed, because in other studies, the incidence of NF1 deletions in CBF-AML did not exceed 2%79,80 and was only 4% in the subset of inv(16) AML.79 In t(8;21) AML with additional del(9q), the putative tumor suppressor genes TLE1 and TLE4 are regularly deleted. In vitro experiments using the human Kasumi-1 cell line, which carries the t(8;21) abnormality, implicated a possible role of TLE-1 and TLE-4 in the development of t(8;21) AML with additional del(9q). The knock-down of TLE-1 and TLE-4 by shRNA in Kasumi-1 cells was associated with increased cell division, whereas the overexpression of both genes resulted in increased cell death and apoptosis.81
In independent studies, among secondary chromosome aberrations in inv(16) AML, only trisomy 22 was found as a favorable prognostic factor that was associated with superior RFS,7,39 lower cumulative incidence of relapse,6 and longer OS.3,39 The results on the prognostic relevance of del(9q) and loss of Y chromosome in t(8;21) AML are discordant among the studies.3,6,7,24
In sporadic AML, the MN1 gene on chromosome 22 is involved in the translocation t(12;22)(p13;q12). Moreover, MN1, which is found to be consistently overexpressed in inv(16) AML, can act as a cooperating hit in the leukemogenesis of inv(16) AML.82 In a mice study, the transplantation of BM cells overexpressing MN1 into irradiated mice resulted in the rapid development of a fatal myeloproliferative disease, whereas mice receiving transplantations with Cbfb-MYH11 chimeric BM overexpressing MN1 developed AML.82 The nature of MN1 as a strong oncogene has been demonstrated more recently in another murine study, where the coexpression of MN1 with MLL-ENL significantly reduced the latency for development of an aggressive AML phenotype compared with MN1 or MLL-ENL alone.83 In the same study, in vitro experiments with human leukemic cell lines overexpressing MN1 showed that siRNA-mediated down-regulation of MN1 expression impaired proliferation, suggesting MN1 as important factor for proliferation.83 In several independent retrospective studies performed in the subset of cytogenetically normal AML, higher MN1 expression was established as an unfavorable prognostic factor.84-86 Whether MN1 expression level affects prognosis in inv(16) AML still has to be determined.
A RUNX1-RUNX1T1 protein variant lacking the c-terminal RUNX1T1 part is translated from an alternative transcript (AML1-ETO9a). The premature truncation appears to bestow the fusion protein higher leukemogenic properties compared with the full-length RUNX1-RUNX1T1 fusion.87 The clinical relevance of AML1-ETO9a expression level is not well established. One Asian study reported that high AML1-ETO9a expression was associated with higher KIT expression, KIT mutations, and worse EFS and OS.88 However, the data from this study need to be confirmed in independent studies on well-defined cohorts with t(8;21) AML.
Monitoring of minimal residual disease
RT-PCR allows a sensitive detection of leukemia-specific RUNX1-RUNX1T1 or CBFB-MYH11 transcripts. In CBF-AML, quantitative PCR-based monitoring of minimal residual disease (MRD) might become useful for patient management. In an AMLSG study on 53 patients with inv(16) AML, relevant MRD checkpoints during the treatment and follow-up period were established, which allowed the identification of patients with high risk for relapse.89 The results from this study showed that repetitive PCR negativity during consolidation and during the early follow-up period was associated with low relapse incidence and that conversion from PCR negativity to positivity after consolidation was associated with a high risk of relapse.89 A subsequent MRC study on 163 patients with t(8;21) AML and 115 patients with inv(16) AML also demonstrated that MRD monitoring at specific time points during the treatment and follow-up is capable of identifying patients at high risk for relapse.90 In that study, MRD threshold levels during follow-up in BM and peripheral blood (PB) were established, which allowed an accurate prediction of relapse in all patients exceeding these MRD values90 ; in t(8;21) AML, the critical threshold levels were more than 500 transcript copies in BM or more than 100 copies in PB, and in inv(16) AML, more than 50 copies in BM or more than 10 copies in PB.90 More recently, a French study prospectively assessed MRD kinetics in BM in CBF-AML [t(8;21), n = 96; inv(16), n = 102] in the context of coexisting gene mutations (KIT, FLT3, NRAS, KRAS).23 In that study, patients were randomized between a standard and a reinforced induction and no difference in RFS was observed between the 2 induction arms.23 In univariable analyses, the presence of a KIT or FLT3 mutation (P = .019; HR = 1.8) and MRD reduction ≥ 3-log between diagnosis and the beginning of the second consolidation (MRD2) were revealed as relevant factors for RFS (P < .001; HR = 0.34).23 Patients with KIT mutations less frequently achieved an MRD2 reduction ≥3-log than patients with KIT wild-type (P = .02; 54% vs 75%).23 In multivariable analysis, MRD2 reduction ≥3-log remained a significant favorable factor for relapse (P < .001; HR = 0.31); the presence of KIT or FLT3 mutation was in trend an unfavorable factor for RFS (P = .11; HR = 1.62).23 The design of this study allowed an allogeneic SCT for patients who failed to achieve MRD2 reduction ≥3-log; however, only 12 (23%) of 52 patients fulfilling this criterion received transplantations in CR1.23 Recent data from a Chinese study on 116 AML patients with t(8;21) investigated the role of MRD for clinical decision making in patients achieving CR1.91 In that study, patients who did not achieve a > 3-log MRD reduction after the second consolidation cycle (major molecular remission) or lost the major molecular remission within 6 months were considered to be at high risk for relapse and, per protocol, were candidates for an allogeneic SCT.91 Although this study suffers from some limitations, high-risk patients undergoing allogeneic SCT had a lower cumulative incidence of relapse (P < .0001; 22% vs 79%) and both superior DFS (P = .001; 62% vs 20%) and OS (P = .007; 72% vs 27%) than high-risk patients treated with chemotherapy alone.91 In contrast, in patients classified as low risk based on MRD kinetics, allogeneic SCT in CR1 did not result in any outcome benefit.91
Summary
There is growing evidence for the genetic heterogeneity of CBF-AML. In addition to the known secondary chromosome aberrations, gene mutations affecting RAS, KIT, and FLT3 have been found to be the most frequent molecular alterations in CBF-AML. It is likely that studies using next-generation sequencing will uncover additional molecular alterations and will provide further insights into CBF leukemogenesis. Genetic markers may become useful to predict outcome in CBF-AML and may serve as novel therapeutic targets, particularly for patients who likely will not to be cured by chemotherapy alone. Indeed, small molecules targeting the highly expressed and frequently mutated KIT or FLT3 kinases have already been integrated into trial concepts in combination with conventional chemotherapy. Because questions related to novel therapeutic approaches need to be answered within clinical trials, it is recommended that CBF-AML patients participate in such clinical trials. The current data do not yet support the routine use of TKIs as an adjunct to chemotherapy in CBF-AML outside of clinical trials. Patients who are not treated on clinical trials, but are eligible for intensive therapy, should receive as frontline treatment a cytarabine- and anthracycline-based induction therapy and HiDAC for consolidation. Whenever possible, older CBF-AML patients should be offered an intensive induction and age-adjusted HiDAC consolidation therapy. Based on the data we have so far, allogeneic SCT should not be generally offered as frontline treatment to CBF-AML patients. Moreover, it is unclear whether patients with CBF-AML and KIT or FLT3 mutations benefit from allogeneic SCT in CR1; this question would also need to be investigated in clinical trials. Because the presence of mutated KIT does not currently influence our primary patient management, KIT mutation screening does not need to be performed routinely in CBF-AML at diagnosis. In contrast, data on KIT and FLT3 mutation status should be collected within clinical trials, particularly in the context of the additional use of TKIs. MRD analysis in CBF-AML patients is strongly recommended at baseline, after each treatment cycle, and every 3 months during the follow-up. When an impending relapse is suspected based on the MRD kinetics, patients should be monitored more closely and the availability of a HLA-compatible donor precautionary explored. However, there are currently no sufficient data supporting preemptive therapeutic interventions in CBF-AML based on the MRD course. Allogeneic SCT is currently considered as a salvage option for relapsing CBF-AML patients.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (grant DO-704-3-1) and the Deutsche Krebshilfe (grant 109675). The authors thank Richard F. Schlenk for his support with creating the figures in this manuscript and Hartmut Döhner for critically reviewing the manuscript.
Disclosures
Conflict-of-interest disclosure: The authors declare no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Peter Paschka, MD, Department of Internal Medicine III, University Hospital of Ulm, Albert-Einstein-Allee 23, D-89081 Ulm, Germany; Phone: 49-731-500-45746; Fax: 49-731-500-45505; e-mail: peter.paschka@uniklinik-ulm.de.
