
Abstract
Hemophilia is caused by a functional deficiency of one of the coagulation proteins. Therapy for no other group of genetic diseases has seen the progress that has been made for hemophilia over the past 40 years, from a life expectancy in 1970 of ∼ 20 years for a boy born with severe hemophilia to essentially a normal life expectancy in 2013 with current prophylaxis therapy. However, these therapies are expensive and require IV infusions 3 to 4 times each week. These are exciting times for hemophilia because several new technologies that promise extended half-lives for factor products, with potential for improvements in quality of life for persons with hemophilia, are in late-phase clinical development.
Historical perspective
Hemophilia is caused by a functional deficiency of one of the coagulation proteins and can lead to spontaneous internal bleeding, which can result in joint damage, intracranial hemorrhage, and death. Hemophilia was documented as a sex-linked disorder more than 1700 years ago in the Talmud.1 In the early 1800s, Otto described the genetics of hemophilia A as an X chromosome-linked bleeding disorder.2 Transfusion of whole blood was shown to successfully treat a hemophilia-associated bleeding episode in 1840.3 The disease gained notoriety because Queen Victoria, who reigned from 1837 to 1901, transmitted hemophilia to the Spanish, Russian, and Prussian royalties. In 1904, Tsarevich Alexis was born as the first male heir to a reigning Russian tsar since the 17th century. After hemorrhages appeared in Alexis, his mother, Empress Alexandra, turned to Rasputin, who was reputed to create miracles, for help. Although thought to be the more common factor VIII (FVIII) deficiency, it was recently found posthumously that Queen Victoria had factor IX (FIX) deficiency.4
The modern era of hemophilia treatment began with the detection of FVIII in human plasma in 19115 and the description of its role in hemostasis in 1937.6 With increasing mechanistic insight into blood coagulation, replacement became more sophisticated, first with the use of plasma in the 1940s, then the development of plasma concentrates in the 1950s, the fractionation of cryoprecipitate in the mid-1960s, and finally the preparation of freeze-dried FVIII that was suitable for storage and use at home in 1968. The availability of factor replacement led to marked improvement in the life expectancy of a boy born with severe hemophilia, from ∼ 20 years in 1970 to essentially a normal life expectancy today.
Along with these advances, it was noted that the mixing of plasmas from 2 different hemophilic patients would occasionally correct the blood clotting defect, which led to the discovery of 2 different defects in most cases of hemophilia, now known as hemophilia A and hemophilia B. Whereas in vitro clotting of plasma consumed the factor deficient in hemophilia A (FVIII), most of the factor deficient in hemophilia B (FIX) was not consumed. The 2 factors were separated because FIX selectively bound to insoluble barium salts, which led to the isolation of the proteins for determination of their partial amino acid sequences. From the protein sequence, reverse genetics was applied to isolate the human genes in the early 1980s and the development of mAbs that were used to produce affinity-purified products. The prevalence of hemophilia A is ∼ 5× that of hemophilia B, which approximately correlates to the difference in size of the 2 X-chromosome–linked genes that serve as targets for mutation and inactivation.
Concerns over virus contamination were heightened when individuals receiving pooled plasma-derived products became infected with hepatitis in the 1970s. Then, in the early 1980s, it became apparent that HIV had contaminated the blood supply because the majority of individuals with severe hemophilia in the United States became infected with the virus. The disastrous epidemics of viral contamination prompted the rapid development of recombinant-derived FVIII, with the first 2 products approved by the Food and Drug Administration (FDA) in 1992, and of recombinant-derived FIX, which was approved in 1997.
Through these developments over the past 50 years, the clinical management for hemophilia has improved dramatically. Protein replacement therapy has reduced the morbidity, improved the quality of life, and normalized life expectancy. Long-term prophylactic therapy reduces or prevents the development of hemophilic arthropathy, is the standard of care for children, and is increasingly being applied to adult care.7-10 The development of recombinant factors has provided a safe and reproducible source for the factors and increased the supply, but these therapies are expensive: costs have risen to > $250 000 per adult patient in the United States. Although prophylaxis is the recommended standard for treatment, these rigorous regimens, often requiring IV infusions every other day, are difficult and adherence remains a problem. In addition, convenient access to peripheral veins remains difficult, and many children require use of central venous access devices, which are associated with the medical risks of sepsis and thrombosis. Importantly, ∼ 25% to 30% of hemophilia A patients and ∼ 3% to 4% of hemophilia B patients develop inhibitory alloantibodies to the infused product.11 Despite recent promising success in gene therapy for hemophilia B,12 a cure for hemophilia is not yet available.
New products in clinical development
The recent years mark the most revolutionary developments in hemophilia since the approval of recombinant factors 20 years ago because several new products are in clinical trials and promise extended half-life (T1/2) for factor replacement therapies. Since the development of recombinant-derived products in the 1990s, there has been an ever-growing interest in developing new products through biotechnology to circumvent present limitations for therapy. A variety of approaches have evolved to increase the T1/2 of the proteins in plasma to reduce dosage frequency with the goal of reducing cost. In addition, with increased structural and functional understanding of the mechanism by which FVIII and FIX exert their coagulant activity, specific variants have been engineered with improved properties. Similar approaches have been applied to activated factor VII (FVIIa) for the treatment of patients with inhibitory antibodies. These products have the potential for simpler prophylactic regimens, obviate the need for central catheters, and possibly improve the quality of life of those afflicted with hemophilia. This review primarily focuses on those therapeutics that have demonstrated potential, or may in the near future, to provide therapeutic advances (Table 1).
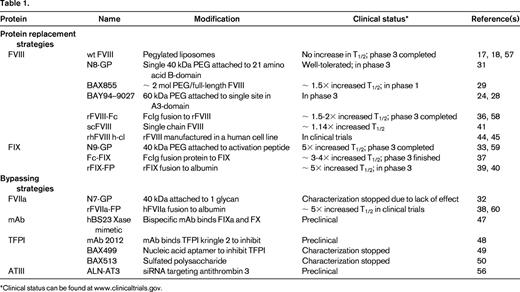
Several approaches have proven successful in increasing the T1/2 of clotting factors in the plasma. There have been modifications to the molecule to reduce proteolytic degradation and modification of the protein.13-15 Initial attempts to mix FVIII with pegylated liposomes as a mechanism for sustained release did not yield significant beneficial results in vivo16 -17,18 Most recent success has been achieved by conjugation at specific sites of the factor with polyethylene glycol (PEG), a hydrophilic polymer, or by fusion of the factor with the Fc fragment of Ig or albumin. Although these fusion approaches have substantially increased the plasma T1/2 of vitamin K dependent factors FIX and FVIIa, less success has been observed with FVIII. This is likely due to different mechanisms of clearance between FIX/FVIIa and FVIII. Previous studies demonstrated that FIX clearance is likely mediated through interaction of the aminoterminal Gla domain residues 3-11 with endothelial/collagen IV sites, although the specific mechanism remains unclear.19 In contrast, clearance of FVIII is very rapid in the absence of VWF, possibly due to binding to phospholipid surfaces of cells. In the presence of VWF, FVIII is stabilized in the plasma to a T1/2 of ∼ 12 hours. The clearance of FVIII from the plasma is likely dependent on the dominant role that VWF plays in regulating the clearance of FVIII.
PEGylation
PEGylation involves the covalent attachment of PEG to a protein to improve its therapeutic effect. In the late 1970s, random PEG addition was shown to reduce the immunogenicity of proteins.20 The most common method of PEG addition is through covalent attachment to lysine residues or N-terminal amines, but this often reduces activity of the protein and the extent of PEG addition is variable, producing a heterogeneous product and complicating reliable synthesis for consistent effectiveness. A new approach involving targeted, site-specific PEGylation has significant advantages.21 One approach to site-directed PEGylation is attachment of PEG-maleimide to cysteine residues. More than 10 PEGylated protein therapeutics have been approved for use, including anti-TNFα mAb Fab fragment,22 VEGF-aptamer, Epoetin β, and IFN-α 2α.23 So far, no long-term safety concerns due to the PEGylation have arisen with any of the approved therapeutics. Of course, weekly use of PEGylated FVIII from early childhood may be a somewhat different clinical setting and, due to the lack of proven clinical experience in hemophilia, appropriate clinical observation will be needed as the clinical trials progress. There are several PEGylated coagulation factors that are in clinical development. In hemophilia animal models of bleeding, all of these PEGylated FVIII molecules have full coagulation activity, and in early clinical trial experience, there are no indications of adverse events, toxicity, and/or immunogenicity in humans. Available safety information of PEGylated proteins containing high-molecular-weight PEG does not indicate any safety concerns to date after long-term (chronic) use in animal models or patients. However, there are differences in how the PEGylation is achieved, and subtle differences may become apparent as the clinical trials proceed through phase 3.
Small PEG molecules are more rapidly cleared than large ones. The rate of excretion is closely related to T1/2. Larger PEG molecules do not permeate into tissues as well as smaller ones. With > 10-kDa PEG, there is increased pinocytotic uptake into macrophages and Kupffer cells; with > 30-kDa PEG, the kidney clearance decreases; and with > 50-kDa PEG, liver clearance increases.24,25 Because all of the PEGylated FVIII products in clinical trials currently use PEG adducts > 15 kDa, specific toxicology experiments for each will need to be evaluated.
PEGylated clotting factors
Several PEGylated coagulation proteins for hemophilia A and B are under development with the goal of prolonging the circulation T1/2 of FVIII or FIX. In one approach, a series of site-directed PEGylated FVIII variants were developed by introducing missense mutations at surface residues on FVIII to incorporate cysteine residues for conjugation with PEG-maleimide. This approach allows selective modifications that do not interfere with FVIII function/activity and also permits greater homogeneity for the final product. This series showed that the PEGylation was highly specific and that the site of PEGylation on the FVIII molecule was critical to PEGylation efficiency, preservation of the coagulation activity, and improvement of pharmacokinetic parameters. In considering cysteine conjugation for FVIII, there are 23 cysteine residues in full-length FVIII, with 3 in the B-domain. Sixteen cysteines are involved in disulfide bonding and 3 are free cysteines, which are likely buried based on the structural analyses.26,27 Through analysis of surface residues mutated to cysteine, it was possible to individually PEGylate each of the 5 domains in B-domain–deleted FVIII.28 PEGylated FVIII retained full activity in vitro and binding to VWF. In addition, the molecule was resistant to inhibition by antibodies directed against the PEGylated epitope. PEGylated FVIII displayed improved pharmacokinetics in mice and rabbits: up to a 2-fold increased terminal T1/2 in a molecule with a 60-kDa PEG addition to Phe129Cys in the A1-domain. Addition of 2 PEG residues in the A1 and A3 or the A2- and A3-domains slightly increased the T1/2 further.28 There was no toxicity associated with acute high-dose administration of BAY 94-9027 or upon repeated dosing with up to 11 mg/kg every other day for 4 weeks in rats. The PEG amount in BAY 94-9027 is ∼ 4 μg/kg in a dose of 60 IU/kg rFVIII. Over a 1-year period, a 50-kg patient would receive ∼ 11 mg of PEG. Although most experts would consider PEG to be inert in the human body, the repetitive and lifelong use of these PEG proteins requires further testing for potential unexpected adverse effects.
Interestingly, the PEG modification at L491C in the A2-domain reduced inhibitory activity toward a mAb that reacts to this highly immunogenic region of FVIII, suggesting that such modifications may limit the inhibitory activity of inhibitory antibodies.28 Bayer Corporation is well into phase 3 studies for the single-site 60-kDa PEGylation mutant B-domain–deleted FVIII K1804C (BAY 94-9027). Several studies have found that PEGylated proteins in general show reduced immunogenicity compared with their unmodified parent molecules.23,24,29 Preclinical studies with the BAY 94-9027 molecule similarly showed consistently less neutralizing antidrug antibody development in rats, hemophilia A mice and rabbits.30 In vitro studies indicated that PEGylation decreased presentation of the rFVIII to APCs, thereby potentially reducing the immunogenicity of FVIII itself compared with unmodified FVIII. Finally, the efficient PEGylation facilitates consistent conjugation at a single site, although thorough characterization and reproducibility of the final product is essential and remains challenging due to the variability in PEG sources. On the positive side, production of these site-directed PEGylated FVIIIs is scalable. It should be noted that PEG is also present in some plasma-derived FVIII products that have been used in patients for many years without safety concerns.
Recently, Baxter International reported a summary of toxicology and preclinical results for BAX 855, a full-length rFVIII PEGylated by chemical means at specific lysine residues. Assessment of animal toxicity was based on mortality, clinical observations, clinical pathology, male fertility in rats, organ weights, and pathology evaluations. No PEG-related effects were observed.29
In a phase 1 clinical trial, Novo Nordisk demonstrated a glycoPEGylated FVIII that increased plasma T1/2 by 1.6-fold. Twenty-six patients each received one dose of their previous FVIII product followed by the same, single dose of the glycoPEGylated FVIII (N8-GP). The clearance of N8-GP was 1.79 mL/h/kg body weight. The estimated time from dosing of 50 U/kg N8-GP to a plasma activity of 1% was 6.5 days (range, 3.6-7.9). The mean terminal T1/2 of N8-GP was 19.0 hours (range, 11.6-27.3), 1.6-fold longer than that of the patients' previous products. A single dose of up to 75 U/kg N8-GP was well tolerated in patients with hemophilia A, with no safety concerns.31
Other PEGylated coagulation factors are also currently in clinical development. GlycoPEGylated rFVIIa (N7-GP), which was manufactured by Novo Nordisk through enzymatic monoPEGylation (> 85% monoPEGylated) of N-linked carbohydrate structures on rFVIIa, results in a 40-kDa PEG moiety attached to the rFVIIa protein.32 To determine the safety and pharmacokinetics of a single dose of N7-GP in healthy men, a randomized, placebo-controlled, dose-escalation trial with 5 cohorts (N7-GP dose of 12.5-100 units/kg) was performed. No serious adverse events were reported. No neutralizing antibodies against N7-GP were detected in phase 1. A second recombinant FVIIa analog with 3 amino acid changes in development (NN1731; vatreptacog alfa) was halted in phase 3 clinical trials when alloantibodies that cross reacted with endogenous FVII were detected in subjects after multiple doses.
N9-GP, a recombinant FIX molecule with site-directed glycoPEGylation of a 40-kDa PEG molecule attached to the activation peptide of FIX, has completed phase 1 and is now in phase 3 clinical development. The toxicology program did not identify any PEG-related safety issues. One serious adverse event was reported in one patient as probably being related to N9-GP, a hypersensitivity reaction (nausea, vomiting, paresthesias, facial swelling, and diaphoresis, but no changes in blood pressure or pulse) that occurred during administration of N9-GP in a 25-year-old male patient who had no history of inhibitors nor allergic reactions to his previous plasma-derived FIX product. After the event, the patient continued with his previous plasma-derived FIX product without any complications.33
Fusion proteins
Another strategy to increasing the plasma T1/2 of clotting factors is to covalently fuse them to proteins that have a much longer T1/2. To date, most success has been reported for fusions to the IgG constant region (Fc), which has a T1/2 of ∼ 3 weeks, or to albumin, which has a T1/2 of ∼20 days. The increase in T1/2 of Fc fusion proteins results from their interaction with the salvage neonatal Fc-receptor present on many cell types, including endothelial cells, monocytes/macrophages, and epithelial cells, in addition to the slower renal clearance for larger-sized molecules.34 The Fc-domain folds independently, can improve the solubility and stability of the partner molecule both in vitro and in vivo, and allows for easy cost-effective purification by protein-G/A affinity chromatography during manufacture. All proteins, including Fc-fusion proteins, are internalized into endothelial cells by pinocytosis to form an endosome. In the process, the endosome is acidified to promote binding of the Fc-fusion protein to FcRn. Upon recycling of the endosome to the cell surface, the Fc-fusion protein is released into the circulation under the neutral pH conditions. In this manner, the Fc-fusion protein escapes degradation by the lysosome. This mechanism is proposed to provide the greater T1/2 of IgG in the blood compared with other antibody isotypes.35 Etanercept and romiplostim are examples of very effective long-acting Fc-fusion proteins. Although Fc-fusions are typically expressed as homodimers formed through a disulfide bond, these dimers were not effective for large clotting factors. Fusion of the monomeric form of the IgG1 Fc to human FIX, FVIIa, and B-domain–deleted FVIII by Biogen Idec has demonstrated increases in plasma T1/2. Monomeric Fc fusion to B-domain–deleted FVIII did not alter its specific activity and displayed a 2-fold longer T1/2 in mice and dogs. The increased T1/2 requires the FcRn. The molecule showed a similar efficacy in tail clip injury in hemophilia A mice and corrected the whole blood clotting time (WBCT) in hemophilia A dogs. The fusion protein shows an ∼ 2-fold increase in sustained protection of the WBCT. In a phase 1 human study in 16 severe hemophilia patients, a single dose of FVIII-Fc demonstrated functional activity, with a 1.5- to 1.7-fold increased T1/2 and no adverse effects or development of antibodies.36
Similar approaches to produce monomeric Fc fusions with FIX have demonstrated a greater impact on T1/2 extension. IV infusion of FIX-Fc into mice and dogs demonstrated a 3- to 4-fold increase in T1/2. In mice, the WBCT was twice as long at the same dose of rFIX. The pharmacokinetics and safety of this molecule were studied in previously treated adults with severe hemophilia B. No adverse events or antibodies were detected. The T1/2 was ∼ 56.7 hours, or ∼ 3-fold longer than that of rFIX.37
Another fusion approach to increase the plasma T1/2 is to covalently attach the clotting factor to albumin. Albumin linkage to coagulation factors FVIIa and FIX (rVIIa-FP and rIX-FP, respectively) increases the plasma T1/2 and the proteins display similar hemostatic efficacy compared with the respective recombinant factors. Clinical evaluation is in progress by CSL Behring, with encouraging preliminary results for rIX-FP in hemophilia B.38-41 To date, there are no studies reported with FVIII-albumin fusion proteins. It is possible that FVIII fusion to albumin may reduce its activity or the fusion may not increase the T1/2 of FVIII, because the major determinant in FVIII clearance is mediated through VWF.
Novel FVIII molecules
An additional approach toward improving FVIII pharmacokinetics was developed using a unique recombinant single-chain FVIII protein that prevents dissociation of the 2 chains of FVIII. This protein has improved intrinsic stability and a higher affinity for VWF relative to other recombinant FVIII molecules. In native FVIII, the heavy and light chains are held together by a labile metal-ion bridge, which makes the FVIII molecule relatively unstable at neutral pH. The recombinant single-chain FVIII was designed with a covalent bond between the heavy and light chains of FVIII. The higher affinity of the molecule may lengthen the T1/2 of the rFVIII molecule in circulation because the clearance of FVIII is mainly determined by its carrier protein VWF, which binds FVIII and protects FVIII from early proteolysis and clearance. This hypothesis was confirmed in different animal species. The biological activity of recombinant single-chain FVIII was comparable to that of full-length rFVIII in terms of activation by thrombin, thrombin generation assay, and tail-clip bleeding in FVIII-deficient mice. This molecule is now in clinical trials in humans.41,42
Another new FVIII molecule in clinical trials is expressed in a human cell line based on the hypothesis that the immunogenicity of the FVIII is less than for FVIII produced in the currently available mammalian, nonhuman cell lines. Host cell lines used for recombinant protein expression differ in their ability to perform posttranslational modifications. For rFVIII, glycosylation and sulfation are vital for functionality and VWF-binding affinity.43 The posttranslational modifications of a novel, human cell line–derived recombinant human FVIII (human-cl rhFVIII) avoids expression of undesirable mammalian glycoforms such as Galα1-3Galβ1-GlcNAc-R (α-Gal) and N-glycolylneuraminic acid (Neu5Gc), which constitute epitopes antigenic to humans. In a recent study, the human-cl rhFVIII was sulfated and glycosylated comparably to human plasma-derived FVIII and was devoid of the antigenic Neu5Gc or α-Gal epitopes observed in Chinese hamster ovary and baby hamster kidney–derived rFVIII products.44,45 Both the avoidance of nonhuman glycan structures and the achievement of complete sulfation are proposed to lower the intrinsic immunogenicity of human-cl rhFVIII compared with current rFVIII products. The clinical trials in progress should provide more information on these novel treatments.
Implications for immunogenicity of novel molecules: a warning
A major concern in hemophilia A is the development of neutralizing antibodies (inhibitors) that prevent further use of therapeutic FVIII. The current options are immune tolerance therapy which is successful ∼ 70% of the time, and the use of bypassing agents such as activated prothrombin complex concentrate or activated FVIIa, both of which are very expensive and less effective at preventing spontaneous bleeding. Therefore, there are significant potential implications as the clinical trials with the new therapies develop. It may prove clinically important that FVIII produced by a human cell line will have a lower rate of inhibitor development. It may be discovered that one or more of the extended T1/2 products will have a lower inhibitor rate. These issues will only be resolved over the coming years through careful monitoring for inhibitor formation in the current and planned clinical trials.
Recombinant-derived FVIIa is safe and effective for treatment of bleeding episodes in hemophilia patients with inhibitors, and FVIIa infusion has not produced neutralizing antibodies in hemophilia patients, probably because they have native FVIIa and do not recognize the infused FVIIa as foreign. In addition, daily prophylactic administration of FVIIa demonstrated significant reductions in bleeding episodes compared with on-demand therapy without significant safety concerns.46 However, its short plasma T1/2 requires short interval follow-up dosing and limits application for prophylaxis. With the current patent on NovoSeven due to expire in 2015, several pharmaceutical companies have development programs to generate molecularly modified FVIIa molecules with a longer T1/2 and/or with improved efficacy. The 2 programs with the most clinical trial data to date are based on molecular constructs that introduce conservative amino acid substitutions into native FVIIa. After finishing phase 1 studies without any safety issues noted, both clinical programs were terminated, possibly due to reduced efficacy. These early results will need much further investigation for their full explanation, but serve as a possible warning for clinical trials for future mutations of clotting factors.
Novel approaches for hemophilia therapies
Although many approaches to improve hemophilia care currently in clinical development involve replacement of modified versions of FVIII, FIX, and FVIIa, there are also several novel approaches being pursued in preclinical work. These novel approaches are intriguing, but much remains to be determined as they enter clinical development.
Factor Xase complex mimetics
FVIII acts to increase the Vmax and decrease the Km for FIXa-mediated cleavage of FX in the presence of negatively charged phospholipids provided by the surface of activated or damaged cells. Current efforts are directed toward identifying small molecules to replace the FVIII dependence by promoting the assembly of FIXa and FX on a negatively charged phospholipid surface in a manner that stimulates the rate of FXa generation. The availability of such a molecule would dramatically affect hemophilia care by providing agents that could be delivered subcutaneously or even orally. A humanized bispecific mAb to FIXa and FX was derived (hBS23) that displayed a 2-week T1/2 in a cynomolgus monkey model of acquired hemophilia.47 The major advance here is the demonstration that it is possible to alter the conformation of FIXa and FX to form an FVIIIa-independent factor Xase activity. This suggests that small molecules with similar potential that could be delivered orally may be engineered.
Inhibition of antithrombotic pathways
The tissue factor/FVIIa/FXa complex forms small amounts of thrombin to initiate coagulation. Tissue factor pathway inhibitor (TFPI) inhibits this complex by interaction of its 2 Kunitz domains. Kunitz domain 1 interacts with FVIIa and Kunitz domain 2 interacts with FXa. A mAb to the second Kunitz domain neutralizes the inhibitory effect of TFPI on extrinsic pathway activation. Subcutaneous administration of this mAb (mAb 2021) 24 hours before injury prevented bleeding in hemophilic rabbits.48
Recently, several therapeutic agents that inhibit TFPI have been described as potential hemophilia treatments. One is a nucleic acid aptamer that binds tightly and specifically to TFPI and inhibits its function in vitro and in vivo.49 Other approaches use a non-anticoagulant sulfated polysaccharide (NASP) BAX 513 that demonstrated efficacy in hemophilia dogs.50 TFPI-antagonist peptides49,51-53 have been designed and shown to have hemostatic efficacy in vitro and in vivo in mouse models. Each type of inhibitor of TFPI provides unique advantages and interactions with clotting factors.53 There is some concern that inhibition of TFPI by certain inhibitor types may worsen bleeding tendencies. Further studies in animals and humans will be needed to sort out the therapeutic opportunities of this potentially exciting approach.
Targeting antithrombin
In another novel approach Alnylam Pharmaceuticals is developing ALN-AT3, a synthetic, GalNAc-conjugated RNAi therapeutic designed to suppress liver production of antithrombin (AT) mRNA when administered via subcutaneous injection. Reducing AT levels has the potential to reduce the stoichiometric inhibition of thrombin and thus improve hemostasis for patients with impaired hemostasis. ALN-AT3 is being developed for the treatment of patients with hemophilia A and B. In pharmacology studies, subcutaneous administration of ALN-AT3 resulted in dose-dependent and reversible reduction of circulating AT, with a single-dose ED50 of ∼1 mg/kg in multiple species.54 AT reduction was associated with significant increases in thrombin generation and enhanced hemostasis in hemophilia A and B mouse models. In a microvessel laser injury model used to compare stable thrombus formation in hemophilia A and B mice, functional improvements in hemostasis were observed upon treatment with ALN-AT3. Animals administered a single dose of 1 or 30 mg/kg ALN-AT3 10 days before injury showed dose-dependent accumulation of platelets and fibrin at the site of injury, as quantified by intravital microscopy. Stable hemostatic plug formation was observed at all sites of injury in treated animals, whereas no stable hemostatic plugs were observed in any of the injury sites in untreated animals.
Extensive toxicology studies have been conducted in several species to support clinical development of ALN-AT3. As expected, the toxicity observed in wild-type animals is primarily attributable to the exaggerated on-target, procoagulant pharmacology of ALN-AT3. Specifically, > 90% AT reduction in wild-type animals typically resulted in adverse thrombotic events and disseminated intravascular coagulation. In contrast, exaggerated exposures of ALN-AT3 (doses up to 500 mg/kg and > 95% reduction in AT) were well tolerated in hemophilia A and B mice, with no evidence of thrombosis, which is consistent with the therapeutic hypothesis of rebalancing an impaired hemostatic system and demonstrate an expanded therapeutic index in the disease condition. Further studies are required in models that have functional FVIII and/or FIX to demonstrate long-term safety.
Conclusion
The past quarter-century has witnessed a tremendous expansion in our understanding of mechanisms that regulate hemostasis in vivo. These advances, together with greater knowledge of the mechanisms that define protein clearance in the plasma, have led the way to several clinical trials for potentially improved clotting factor products for persons with hemophilia A or B. New extended T1/2 therapeutics may allow less frequent infusions of factor, allow higher factor trough levels so there may be increased potential for preventing spontaneous bleeding, and may be associated with a lower inhibitor rate. In addition, there may be alternative approaches to preventing bleeding in hemophilia that will soon be entering clinical trials. Future questions include: (1) how will the costs of these new factor products influence their clinical use; (2) will there be any issues with long-term tolerance with modified factor products; (3) what will be the experience with developing inhibitors after use of these new factor products in previously untreated patients; and (4) will there be any new issues of concern in using these new factor products in an increasingly elderly hemophilia population? Enthusiastic participation by physicians and patients will allow these clinical trials to proceed and obtain appropriate answers to these significant questions for the hemophilia community.
This article was selected by the Blood and Hematology 2013 American Society of Hematology Education Program editors for concurrent submission to Blood and Hematology 2013. This article is reprinted with permission from Blood. 2013; Volume 122.
Acknowledgments
This work was supported by the National Institutes of Health (grants HL057346 and HL052173 to R.J.K.). The authors thank Glenn Pierce for insightful and constructive comments during the final stages of preparing this review and Jorg Schauttrumpf (German Red Cross Blood Donor Service), Rod Camire (Children's Hospital of Pennsylvania), Camilla Brock (Bayer), Stefan Lethagen (Novo Nordisk), and Rachel Meyers (Alnylam Pharm) for providing information and advice. The authors apologize to those whose studies were omitted due to space limitations.
Disclosures
Conflict of interest disclosure: J.S.P. has received clinical trial support from Biogen, Bayer, Baxter, Octapharma, CSL Behring, NovoNordisk, and GTC. R.J.K. declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Randal Kaufman, Sanford Burnham Medical Research Institute, 10901 North Torrey Pines Road, La Jolla, CA 92037; Phone: 858-795-5134; Fax: 858-795-5279; e-mail: rkaufman@sanfordburnham.org.
