
Abstract
Diffuse large B-cell lymphoma, the most common lymphoma subtype, is curable in the majority of patients. However, one of the greatest unmet needs in lymphoma treatment remains novel approaches to prevent relapsed or refractory disease. Genomic profiling has provided important prognostic information that is being used in the development of novel therapeutic strategies currently in clinical trials. It is clear, however, that epigenetic alterations provide an additional series of targets that can be pharmacologically modified and offer great potential to improving patient outcomes. Greater understanding of this area is providing important new insights that are now being explored in the clinical setting. Demethylating agents and drugs that disrupt histone modifiers are in early clinical trials with promising results, and other approaches targeting epigenetic pathways are in active preclinical and early clinical development.
Selected novel approaches to target the epigenome in diffuse large B-cell lymphoma
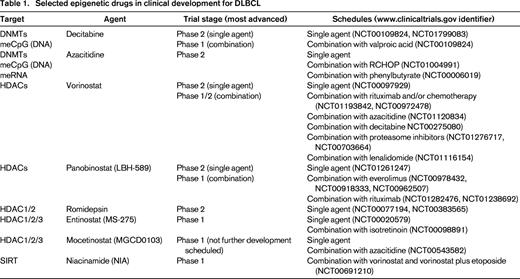
Genome-wide DNA methylation and histone modification patterning and next-generation sequencing leading to directed functional studies have vastly increased our understanding of epigenetic deregulation in diffuse large B-cell lymphoma (DLBCL). Most importantly, these analyses have revealed rational therapeutic targets for development (Table 1). Epigenetic alterations in lymphoma, in contrast to genetic lesions, are themselves pharmacologically reversible and therefore represent strategies for novel therapeutic approaches. In the clinical setting, epigenetic therapy is currently limited to pharmacologically tilting the balance in favor of histone acetylation and/or DNA hypomethylation to cause antilymphoma effects. Histone acetylation relaxes chromatin, which leads to transcription activation and reexpression of genes that can result in favorable biologic responses, including apoptosis of tumor cells. In addition, DNA demethylation can induce the reactivation of genes that are silenced by hypermethylation, causing similar biological effects that can result in tumor cell death. Several examples of epigenetic therapy approaches for DLBCL are currently being used in the clinical setting or are expected to be explored in patients in the near future.
Targeting aberrant DNA methylation
DNA methylation patterning contains epigenetic information that encodes transcriptional programming information that leads to the phenotype of normal and malignant cells.1 Aberrant DNA hypermethylation of tumor suppressor genes can result in their inappropriate transcriptional silencing, which contributes to loss of checkpoints and related functions in cancer. Inactivation of tumor suppressor pathways is therefore an important contributor to resistance to chemotherapy in cancer,2-4 in part because the activity of most chemotherapy agents largely depends on the same proapoptotic and prodifferentiation pathways that become disabled during carcinogenesis. Inactivation of these pathways by mutations or hypermethylation can therefore affect drug sensitivity.2,5 In addition, epigenetic inactivation of chemotherapy drug transporters and cellular damage repair mechanisms can also result in chemoresistance. Oncogene expression can also be a consequence of DNA methylation. For example, BCL6, which contributes to lymphomagenesis in the germinal center DLBCL subtype, is maintained in lymphomas in part through DNA methylation that prevents CTCF-mediated gene silencing.6 Gene-specific and genomic alterations in DNA methylation have been described in various subtypes of non-Hodgkin lymphoma (NHL).7-13 Moreover, genome-wide analysis of DNA methylation in DLBCL and follicular lymphoma (FL) specimens suggests that aberrant DNA methylation increases with disease aggressiveness.14 The highest extent of DNA methylation deviation from normal tissues was described in the more chemorefractory (and poorer prognosis) activated B-cell-like DLBCL subtype.14 Importantly, these abnormal methylation patterns are not randomly distributed, but rather are associated with specific genomic regions that depend on the activity of target-specific transcriptional regulators (such as BCL6 or EZH2), as well as the absence of insulators such as CTCF that allow the spread of hypomethylation to neighboring genes.6,14 These studies suggest that epigenetic abnormalities provide a survival advantage for lymphoma tumor cells and that such clonal epigenetic diversity and evolution can ultimately lead to more aggressive and chemoresistant disease.
DNA methyltransferases (DNMTs), including DNMT1, DNMT3A, and DNMT3B, can methylate DNA specifically at cytosines in CpG dinucleotides. DNMT1 is predominantly involved in maintenance, whereas DNMT3A and DNMT3B primarily mediate de novo cytosine methylation. Although there do not appear to be recurrent mutations in DLBCL that affect these genes, DNMT1, DNMT3A, and DNMT3B were found to be overexpressed in 48%, 13%, and 45% of de novo DLBCLs, respectively, and correlate with advanced clinical stage.15 Concomitant expression of DNMT1 and DNMT3B is correlated with resistance to treatment, whereas DNMT3B overexpression is associated with shorter overall and progression-free survival.15 These findings suggest that the targeting of such processes through the development of specific DNMT inhibitors could be useful in the treatment of lymphomas.
Inhibition of DNMT activity can reverse DNA methylation and gene silencing and therefore restore expression of important gene pathways.9 5-aza-2′-deoxycitidine (decitabine, Dacogen) and azacitidine (Vidaza) are cytidine nucleoside analogs that incorporate into DNA and irreversibly inactivate DNMT by forming a covalent bond between the 5-azacytosine ring and the enzyme. As a consequence, DNMTs become unable to efficiently introduce methyl groups in newly synthesized DNA strands. This results in the gradual depletion of 5-methyl-cytosines from the genome as cells divide. Another cytidine analog, zebularine, also exhibits weak DNA hypomethylation activity. Zebularine inhibits cytidine deaminase by binding to the active site. Classical DNMT inhibitors (DNMTi's), including decitabine and azacitidine, can induce significant clinical responses and even prolong the survival of patients with higher-risk myelodysplastic syndrome16,17 and are being actively investigated in other myeloid malignances.18 These studies raise the possibility that DNMTi's might be useful in tumors with active DNA replication, such as DLBCL; however, the optimal dose schedule for these agents remains to be fully clarified.
Major challenges for the successful translation of DNMTi's into the clinical setting include many of the usual issues with novel agents, including optimal patient selection, development of pharmacodynamic markers of biologic activity, and establishment of dose and schedule for combinations with chemoimmunotherapy. A phase 1 trial of DNMTi's for lymphoma patients that used a classical approach for anticancer agents (ie, the use of maximally tolerated doses) showed a low therapeutic index.19 With the goal of using DNMTi's in a biologically defined manner, our group at Weill Cornell Medical College has recently investigated the preclinical pharmacology and effects of DNMTi's in DLBCL patients.20 We hypothesized that pretreatment with the DNMTi would result in demethylation and reexpression of genes that play a role in modulation of sensitivity to chemotherapy in lymphoma cells. We found that, consistent with another recent publication,21 doses of DNMTi's that induce DNA demethylation are ∼10-fold lower than those required to induce significant DNA damage. This is a key concept for the clinical translation of DNMTi's because it supports the strategy of drug administration at relatively low doses for a longer time to maximize the exposure of lymphoma cells to the agent. In this regard, the availability of oral version of DNMTi's such as oral azacitidine CC-48622 will likely represent a substantial improvement based on schedule implementation due to additional flexibility in dosing relative to parenteral treatment. Oral administration of hypomethylating agents would be expected to significantly facilitate the delivery of prolonged, subcytotoxic doses that will likely be required for adequate demethylation within lymphoma tumor cells.
In our preclinical studies, we also found that gene demethylation followed by phenotypic changes (reprogramming) is critical to chemosensitization.23 It appears that reactivation of specific pathways (such as TGF-β) may be more relevant therapeutically than achieving global DNA hypomethylation. Exposure of chemotherapy-resistant DLBCL cell lines to prolonged, low-dose DNMTi's resulted in hypomethylation and decreased cellular growth rates, as well as the induction of a senescence-like phenotype that has been shown previously to be associated with increased sensitivity to doxorubicin-induced genotoxic stress. Subsequent treatment of these cells with chemotherapy in a sequential fashion, both in vitro and in vivo, resulted in increased cell killing. Conversely, concurrent DNMTi and doxorubicin administration failed to achieve significant effects compared with each drug alone, suggesting that epigenetic reprogramming drugs may also require adequate time to sensitize tumors to the effects of DNA-damaging agents. In contrast to normal tissues, in which DNMTi's usually induce cellular differentiation, in our studies, reprogramming of DLBCL cells was notably associated with reactivation of the TGF-β pathway (specifically SMAD1), resulting in “tumor suppressor” effects and the appearance of molecular and phenotypic markers of senescence without typical cell cycle arrest.
Based on these preclinical data we conducted a phase 1 clinical trial of subcutaneous azacitidine (administered for 5 days, beginning 1 week before each R-CHOP [rituximab plus cyclophosphamide, hydroxydaunorubicin, vincristine, prednisone/prednisolone] cycle) for 6 cycles of 21 days.24 Core needle biopsies of accessible lymph nodes were performed on days 1 and 8 of cycle 1. A total of 12 patients were treated, all but one of which were over age 60 and had a high-intermediate or high International Prognostic Index score. Dose escalation of azacitidine took place according to a continual reassessment method and 75 mg/m2 (the highest dose evaluated) was established as safe. Two patients experienced dose-limiting toxicity: one reactivation of hepatitis C (likely related to rituximab, less likely to azacytidine) and one gastrointestinal bleed from a responding gastric lymphoma. Grade 4 neutropenia was common and 4 patients experienced grade 3 febrile neutropenia. All but one patient achieved a complete response to treatment and, interestingly, only 2 patients in this poor-prognosis group have progressed so far, although follow-up is ongoing. Biopsy samples taken before and after azacytidine demonstrated global DNA demethylation, reactivation of the TGF-β pathway (up-regulation of SMAD1) after therapy, and increased ex vivo sensitivity to chemotherapy.23 These data demonstrate that subcutaneous azacitidine before R-CHOP chemotherapy is biologically active and clinically feasible in DLBCL patients, providing rationale to further evaluate the ability of such an epigenetic priming approach to improve therapeutic outcomes.
Targeting histone modifications
Another potentially actionable major component of the epigenetic regulation is the so called “histone code.”20 This area is represented by a group of covalent histone protein modifications that regulate the transcriptional machinery. This coding is edited by histone-modifying enzymes that function as “writers,” “erasers,” and “remodelers,” that are interpreted by “readers.” This network of chromatin modifiers plays an essential role in the adaptation of lymphoma cells to environmental and intrinsic cellular conditions. Genetic and epigenetic abnormalities affecting histone-modifying enzymes and regulators such as histone deacetylases (HDACs) have been described in DLBCL, although the consequences for lymphomagenesis or response to chemoimmunotherapy are still not clear. Given the initiation of several clinical trials exploring HDAC inhibitors (HDACi's) for the treatment of DLBCL, mutations affecting HDACs or their functional counterparts histone acetyltransferases (HATs) are of potential clinical interest. Recurrent mutations in HATs, such as CREB-binding protein (CREBBP) and E1a-binding protein 300 (EP300), have been identified in 16% to 39% of DLBCLs or FLs.25-28 Although the balance between HATs and HDACs can be pharmacologically manipulated using HDACi's, preclinical studies suggest that DLBCL with CREBBPHAT and/or EP300HAT mutations may be resistant to this strategy.25 In addition to histones, other proteins, including transcription factors such as p53 and BCL6, are subject to regulation by acetylation. Therefore, HDACi's may have pleiotropic effects in DLBCL and may be better used in combination with biological agents.25,29 Nevertheless, recent preclinical data suggest that a weak HDACi, valproic acid, may sensitize DLBCL cell lines to CHOP-induced cell death.30
Although there are several ongoing and completed trials of HDACi's in DLBCL patients, most of the reported single-agent results showed at best modest effects in recurrent disease. For example, as a single agent in relapsed DLBCL, vorinostat (SAHA) achieved response in 1/18 patients in a phase 2 trial.31 Another hydroxamic acid, panobinostat, is being evaluated alone and in combination with rituximab for relapsed/refractory DLBCL (personal communication from Sarit Assouline, McGill University, www.clinicaltrials.gov identifier NCT01238692). Emerging data from single-agent and combination studies with noncytotoxic agents indicate that HDACi's may have an acceptable toxicity profile that suggests potential feasibility of combination therapy with R-CHOP.
Other epigenetic targets
EZH2 is a member of the polycomb repressive group 2 (PRC2), which is involved in maintenance of the transcriptional repressive state of genes. EZH2 represents the catalytic subunit of the PRC2, which methylates lysine 9 and lysine 27 of histone H3 (H3K9me and H3K27me), leading to transcriptional repression of the target gene. In normal germinal center B cells, DNA methylation and H3K27me3 marks are mutually exclusive, whereas this epigenetic segregation has been shown to be disrupted in DLBCL.32 These aberrant epigenetic events may be due in part to mutations in the SET domain of EZH2 that have been detected in up to 12% of FL and 9.7% of DLBCL cases.33,34 EZH2 inhibitors are expected to be relevant therapeutically for patients with gain-of-function mutant EZH2 and/or germinal center DLBCL.35,36
Combination epigenetic-targeting approaches
Preclinical data suggest that multiple epigenetic mechanisms cooperate for gene silencing.37,38 Moreover, in vitro and in mouse xenograft experiments with the combination of panobinostat and decitabine in DLBCL have shown synergistic effects resulting in increased gene reexpression and superior antilymphoma activity without increased toxicity.39 Similar to combination with chemotherapy agents, the optimal schedule and dosing of this combination has not been fully established to date. A phase 1 study of vorinostat in combination with decitabine that included previously treated NHL and solid tumor patients has explored concurrent and sequential schedules of administration.40 These investigators found that both schedules were equally tolerable, but the sequential administration of decitabine (10 mg/m2/d on days 1-5) followed by vorinostat (starting at 200 mg 3 times a day on days 6-12) was more practical to deliver.40 This also represents the dose level and schedule with the highest percentage of patients with stable disease. Our group is currently evaluating vorinostat in combination with azacitidine in patients with recurrent DLBCL in an ongoing trial.
Lysine acetylation inhibits the activity of BCL6 by inducing its degradation while increasing the activity of p53. HDACs comprise a family of 18 members that are separated into 4 classes. Classes I, II, and IV operate by metal-ion-dependent mechanisms, whereas class III (referred to as sirtuins, SIRT1-7) operate by an NAD+-dependent mechanism. In addition to metal-binding HDACs, SIRT1 is also responsible for BCL6 and p53 deacetylation. Therefore, the activity of the transcriptional factors BCL6 and p53 is controlled by competing lysine acetyl-transferases (KATs) and lysine deacetylases (HDACs and SIRT1). Cambinol, a SIRT1 inhibitor, has been shown to inactivate BCL6 and induce apoptosis in both in vitro and in vivo models of NHL.41 Due to subcellular localization and differential affinity for target inhibition amongst other factors, lysine deacetylase inhibitors may offer distinct therapeutic properties. Therefore, a combination of different HDACi's may deliver a greater antilymphoma effect. The successful combination of vorinostat with the SIRT inhibitor niacinamide has been shown in a phase 1 trial of patients with recurrent HL and NHL.42 The dose-limiting toxicity for this regimen included neutropenia, infection, and transaminitis, and response assessments are ongoing.
Conclusions
Epigenetic pathways represent relevant and promising therapeutic targets in DLBCL. Several agents have entered the clinical setting and have demonstrated pharmacodynamic effects. In addition, recent studies have suggested that the sequential combination of epigenetic priming before R-CHOP is feasible and shows promising clinical outcomes in patients with previously untreated DLBCL. Combination epigenetic therapy has also entered the clinical setting. Ultimately, continuing studies to optimize patient/tumor selection and to establish pharmacodynamic markers of biologic effects will allow further development of these promising therapeutic strategies in combination with standard agents to maximize the benefit for patients.
Acknowledgments
L.C. is a Scholar of the American Society of Hematology and the Raymond and Beverly Sackler Scholar and is supported by the Doris Duke Charitable Foundation and the Irma T. Hirschl Trust. J.P.L. is supported in part by the Lymphoma Foundation and the Cancer Research and Treatment Fund.
Disclosures
Conflict-of-interest disclosure: J.P.L. has consulted for Spectrum, Celgene, Medimmune, Biotest, Sanofi Aventis, Gilead, Onyx, Hospira, Millenium, Pharmacyclics, Johnson and Johnson, and Genentech. L.C. declares no competing financial interests. Off-label drug use: Investigational and off-label lymphoma therapies.
Correspondence
John P. Leonard, MD, Division of Hematology and Medical Oncology, Weill Cornell Medical College, 1305 York Avenue, Rm Y-744, New York, NY 10021; Phone: 646-962-2068; Fax: 646-962-1605; e-mail: jpleonar@med.cornell.edu.
