
Abstract
For children with acute lymphoblastic leukemia, the identification of those at higher risk of disease recurrence and modifying therapy based on this risk is a critical component to the provision of optimal care. The specific definitions of high-risk ALL vary across cooperative groups, but the themes are consistent, being largely based on leukemia biology and disease response. Intensification of conventional chemotherapy for those with high-risk disease has led to improved outcomes. It is anticipated that the development of rational targeted therapy for specific biologically unique subsets of children with leukemia will contribute to ongoing progress in improving the outcomes for children with acute lymphoblastic anemia.
Learning Objectives
To understand current criteria used to identify children with high-risk ALL
To understand therapeutic strategies for children with high-risk ALL
Introduction
The chance of successful treatment for a child or adolescent with acute lymphoblastic leukemia (ALL) has improved dramatically over the last 50 years, with 5-year overall survival (OS) rates of approaching 90% for children receiving therapy in the developed world.1,2 The identification of active chemotherapy agents, understanding how to use them in combination, and treatment of the CSF as a sanctuary site were the steps that moved the disease from one that was uniformly fatal to one with a chance of cure of ∼75% by the 1980s.3-7 The majority of the improvement in outcomes in the last several decades can be attributed to modifications of the use of these original components and, to some extent, to intensifying therapy for those patients identified as having disease that is more difficult to cure. In this brief review, contemporary criteria used to identify patients at higher risk of relapse and the utility of intensification of therapy based on this risk are reviewed.
Risk classifications
Risk classification is needed when there is uncertainty regarding a future event with estimates of the probability of the event being made based on past experience. For the purposes of care of a child with newly diagnosed ALL, the future event is relapse of disease and past experience is largely generated from data from cooperative clinical trials. Risk classification is important for determining the average outcome for a group and for determining whether interventions can modify the risk. Risk classification is not the same as being able to predict an individual's experience.
For the care of patients with ALL, the ideal risk classification system uses factors that are simple to measure and therefore easily generalizable and are able to be determined at the time of diagnosis or early on during the treatment course. Such a system then allows for risk-adapted therapy with treatment modifications made that may affect the likelihood of success.
History of risk stratification in ALL
The importance of understanding why certain patients are cured of their disease and others are not has been an integral part of the development of ALL therapy. Reporting in the Journal of the American Medical Association in 1966, Sidney Farber described 15 long-term survivors from the initial group of 1445 pediatric patients with ALL treated with chemotherapy. He notes, “it is still impossible to differentiate, at the time of diagnosis, the >99% whose lives will be prolonged by months or one or 2 years and the <1% who will survive 5 years of longer.”8 In the 1970s and 1980s, with increasing success of ALL therapy, investigators began to identify risk factors for relapsed disease. Patient age, disease burden measured in presenting WBC or “tumor bulk,” and evidence of disease response measured by changes in peripheral blood blast count or early BM morphologic assessment were found to be predictive of outcome.9-11 In the early 1990s, the 4 cooperative groups running clinical trials in pediatric ALL in North America all used risk definitions that included age and WBC, but all used different cutoff points for these continuous variables. The need to be able to compare and apply results across studies lead to a consensus conference in 1993 leading to the creation of the National Cancer Institute (NCI) criteria defining high-risk patients with B-precursor ALL as those 10 years of age or older or those having a presenting WBC count of 50 000/μL or greater.12
Contemporary strategies
The information used to allocate patients with ALL into different risk groups in contemporary trials can be divided into 3 categories: the patient's clinical features at the time of diagnosis, the disease characteristics, and measurements of initial response to therapy.
Within the Children's Oncology Group (COG), patients with ALL are currently classified into 7 different groups including infants, those with Philadelphia chromosome-positive (Ph+) disease, and those with T-ALL. The classification of the remainder of children with precursor B-ALL into 4 additional groups is described in Table 1. Factors used to define the risk group of children with ALL by other cooperative groups in contemporary clinical trials are similar in themes but vary in their details (Table 2).
COG classification of newly diagnosed B-precursor ALL (adapted from the COG classification system for B precursor ALL, AALL08B1)86

Favorable genetics: hyperdiploidy with trisomies of chromosomes 4 and 10 or ETV6_RUNX1 fusion. Unfavorable characteristics: CNS2, induction failure, age 13 y and older, hypodiploid (<44 chromosomes or DNA index <0.81), MLL rearrangement, iAMP21. Note that infants and those with Ph+ disease are treated on separate trials.
HR, High risk by age and WBC count; PB, peripheral blood; and SR, standard risk by age and WBC count.

Clinical features
Age at diagnosis remains an important predictor of risk of relapse. Excluding infants, older age is consistently associated with a worse outcome, which can be explained in large part by variations in the biology of the disease with age. A smaller proportion of older children and adolescents with ALL have favorable cytogenetic findings, including ETV6-RUNX1 or hyperdiploidy, and a higher proportion have unfavorable genetic features including hypodiploidy and BCR-ABL (Ph+) disease.13 Contributing to the inferior OS of older patients is the observation that those >15 years of age treated for relapsed ALL have a significantly worse outcome than their younger peers.14
Infants, those <1 year of age at the time of diagnosis, remain at very high risk of relapse, even with aggressive contemporary therapy.15,16 Risk stratification within the infant group is based on presence of MLL gene rearrangement, found in ∼75% and in the majority of younger infants. In addition, months of age at the time of diagnosis, with those <3 or 6 months fairing worse and high presenting WBC (>100 000 or >300 000) are important prognostic factors within this group of patients.17
WBC count at the time of diagnosis of >50 000/μL is associated with less favorable outcome for children with B-lineage ALL.12 Age and WBC count are not accurate predictors of outcome for T-cell ALL.18,19
Patients meeting the criteria of CNS 3 (having 5 WBCs/μL in the CSF with blasts in the cytospin) and, at least within the context of some studies, those with initial traumatic lumbar punctures with blasts (≥10 RBCs/μL) have an inferior event-free survival (EFS).20,21 Presence of CNS disease at the time of diagnosis is associated with other adverse prognostic factors.22 The prognostic implication of CNS 2 (<5 WBCs/μL with blasts) has been less consistent but likely also confers higher risk.23,24
Patients with Down's syndrome and ALL have both a higher risk of relapse and a higher rate of treatment-related mortality (TRM) than non-Down's syndrome patients.25 Evaluation of >600 patients across 16 cooperative groups trials suggest that cutoff values for age and presenting WBC count for risk allocation in children with Down's syndrome should not be the same as those used for non-Down's syndrome patients.
Gender and ethnicity have not been used in the allocation of treatment intensity in clinical trials, except for the use of prolonged maintenance therapy for boys in some cooperative group protocols. Boys, irrespective of risk of testicular relapse, have a slightly inferior outcomes compared with girls.26 Black and Hispanic children with ALL have been shown to have an inferior outcome compared with Caucasian and Asian children.27 The reasons underlying these disparities are likely multiple, but include variations in ALL subtypes, host polymorphisms, and sociodemographic factors.28,29
Disease factors
In earlier treatment eras, T-cell phenotype was considered an unfavorable prognostic feature, but over the last decade, with more intensive therapy and, especially if those with NCI standard risk features are excluded, patients with T-cell disease have been shown to have outcomes similar to those with precursor B-ALL.30-32 Unlike precursor B-ALL, prognostic factors for patients with T-cell ALL are less clear. A subset of T-cell ALL patients with a unique phenotype, described as early T-cell precursor ALL, characterized by being CD1a and CD8 negative with weak CD5 and coexpression of myeloid markers, have been shown to have an highly unfavorable prognosis by some groups and an intermediate-risk outcome by others.19,33-35
The list of genetic alterations in childhood ALL that are associated with risk of relapse continues to rise.36 Some of these factors are well established as predictive of outcomes, whereas others remain to be validated.
Patients with hypodiploid leukemic cells have an unfavorable prognosis; those with fewer than 44 chromosomes are at the highest risk.37 A significant portion of ALL samples with low hypodiploidy will have additional unfavorable genetic findings, including TP53 alterations in >90% of cases.38
Patients with Ph+ ALL [t(9:22)] historically had very poor outcomes, especially those with higher WBC and slow early response to therapy.1 A significant portion of patients with Ph+ ALL will have alterations in IKZF1, which is associated with chemotherapy resistance.39 With the introduction of tyrosine kinase inhibitors to intensive conventional therapy, the outlook has changed substantially.
Ph-like or BCR-ABL1-like ALL makes up 10%–15% of pediatric ALL and is identified by having a gene expression profile similar to that of Ph+ disease but without the presence of the BCR-ABL translocation.40,41 It is associated with a high frequency of IZKF1 alterations and is associated with a poor prognosis.42,43 This entity has not been included in risk stratification schemes in clinical trials to date but likely will be in the near future.
In infants with ALL, in whom translocations involving the MLL gene are very common, any MLL rearrangement confers a significantly worse prognosis.15 In older children, the prognostic importance of MLL is somewhat less clear and may be affected by age and translocation partner.44 Some, but not all, cooperative groups use the presence of MLL gene rearrangements as an indication for allocation to higher-risk therapy.
Initial descriptions of patients with intrachromosomal amplification of RUNX1 (iAMP) described very poor outcomes.45 More recent data suggest that intensification of conventional chemotherapy abrogate this risk.
With the availability of newer genomic techniques, multiple recurrent submicroscopic genetic alterations, including gene deletions, mutations, and amplifications, have been identified.46 Many of these alterations have not yet been shown to have clinical meaningfulness. One of the more common of these abnormalities is IKZF1 alterations, which occur in ∼15% of pediatric ALL cases and are associated with an unfavorable prognosis.41,47 IKZF1 alterations are found more commonly in patients meeting NCI high-risk criteria, those with Ph+ and Ph-like disease, and in patients with Down's syndrome.39,48,49
Response factors
Response to therapy as a prognostic indicator has been used since the early trials of ALL therapy. Failure to obtain remission after 4-6 weeks on induction therapy is a relatively rare event, occurring in ∼2%-3% of patients with ALL, and is associated with a poor OS.50 Those with induction failure are more likely to have other high-risk features, including older age and higher WBC, having T-cell disease, and having disease with unfavorable genetic features.
The rate of clearance of blasts from peripheral blood and BM by assessment of morphology has been shown to be prognostic and has been used for risk group allocation in many ALL trials.51,52 Prednisone response as measured in peripheral blood after 7 days of monotherapy and day 8 and day 15 of induction therapy assessments of BM morphology have been critical factors in risk assignment strategies in the recent past. These response tools remain valuable, including their use in contemporary trials in countries with variable resources in which minimal residual disease (MRD) measurements are not routinely feasible.53
For the majority of pediatric ALL subgroups, the measurement of MRD has been shown to provide the most powerful prognostic information. Several recent large clinical trials have cemented this tool as the most important prognostic variable for the majority of children with both precursor B-ALL and T-cell ALL.19,54-56 In a study involving 2143 patients with NCI high-risk and standard-risk ALL, MRD measured by flow cytometry evaluated at the end of induction (day 29) was found to be the most significant prognostic factor, although NCI risk group maintained prognostic significance in multivariate analysis.54 Therapeutic interventions were not made based on MRD in this series of trials. Of the high-risk patients included in this study treated on the POG 9905 regimen, those who had an MRD level >0.01% at the end of induction had a 5-year EFS of 33% ± 8%) compared with 79% ± 4% (p < .001) for those who were negative.
The AIEOP-BFM 2000 study used PCR-based MRD measurements at the end of induction (day 33) and the end of consolidation (day 78) in 3184 patients with precursor B-ALL to prospectively risk stratify patients.55 Those who had MRD levels >10-3 at day 78, as well as those with a prednisone poor response, induction failure, or those with chromosomal translocations of t(4;11) or t(9:22), were allocated to the high-risk group. High-risk patients received intensified blocks of postconsolidation therapy. Even in the context of allocation of more intensive therapy, in multivariate analysis, MRD at the end of induction was the most important factor, although elevated WBC and ETV-RUNX1 status and DNA index retained predictive value. By MRD risk assignment alone, the EFS at 5 years was 91% in the standard-risk group compared with 77% in the intermediate-risk group and 50% in the high-risk group.
In the UKALL 2003 trial, patients were risk stratified based on clinical features, cytogenetics, and early BM response, as well as by MRD evaluation at the end of induction and postconsolidation. MRD risk status, with high risk defined by the presence of 0.001% MRD at the end of induction, was the most significant prognostic factor. Of the patients with evaluable MRD, 81% of all relapses occurred in those who were MRD high risk.56
Within the context of the AIEOP BFM 2000 study, MRD measurement in patients with T-cell ALL was the most powerful prognostic factor for risk of relapse,19 although prednisone poor response and early T-cell phenotype also maintained prognostic value. The kinetics of MRD clearance in patients with T-ALL is different with than in those with precursor B-ALL, with only a small proportion (16%) being MRD negative by day 33. Conversely ∼50% are negative by day 78 and have a 7-year cumulative incidence of relapse of 8.5%. The incidence of relapse of those who are positive at day 78 was significantly higher and varied by level of MRD positivity, with those with >10-3 having an incidence of relapse of 44.7%.
In addition to the traditional factors used to assess risk of relapse for patients with ALL, consideration of the patient's compliance with therapy may need to be taken into account. In a study evaluating adherence with oral 6 mercaptopurine in the maintenance portion of treatment, an adherence rate of <95% was associated with a significantly increased risk of relapse.29
Treatment for high-risk ALL
Identification of groups at variable risk of relapse is done primarily to be able to inform modifications of therapy to limit short-term and long-term toxicities to those with more easily treatable disease and to intensify therapy for those with a worse prognosis. Improvement in outcome for higher-risk patients to date can largely be attributed to intensification of conventional chemotherapy. For a small percentage of children with very-high-risk ALL, intensification of therapy with the use of stem cell transplantation (SCT) in first remission has been applied. The use of biologically targeted therapy for those with less favorable disease to which a druggable target can be identified is clearly the most appealing intervention. To date, the success of this strategy is largely limited to the subset of patients with Ph+ leukemia.
Therapy for ALL is generally divided into 3 components, including remission induction, consolidation/intensification, and maintenance (also referred to as continuation).
Induction therapy for high-risk patients on most contemporary cooperative group trials use at least 4 drugs, including vincristine, an anthracycline, an asparaginase product, and either prednisone or dexamethasone. The most commonly used anthracyclines are doxorubicin or daunorubicin. Compared in a “therapeutic window” design as part of the induction regimen, the 2 agents were found to be equally effective.57 Mitoxantrone was shown to be superior to idarubicin in induction therapy for patients being treated in the relapsed setting, but to date is not commonly used as part of initial therapy.58 Data to support the choice of asparaginase product in induction is lacking, but most contemporary trials use pegylated asparaginase, with IV administration being demonstrated to be safe.59 Dexamethasone and prednisone are both used in induction regimens. With the limitation of variability on conversion ratios used between the 2 agents, data suggest that dexamethasone may be more effective, but is associated with increased risks of acute and long-term toxicities.60
Intensification of therapy for high-risk patients has been done primarily during the 6-8 months of therapy after induction. Randomized trials evaluating postinduction intensification in high-risk patients over the last 2 decades have demonstrated significantly improved outcomes with some intensified regimens, but others showed no benefit (Table 3).
Selected randomized clinical trials evaluating intensification of postinduction therapy for patients with higher-risk ALL
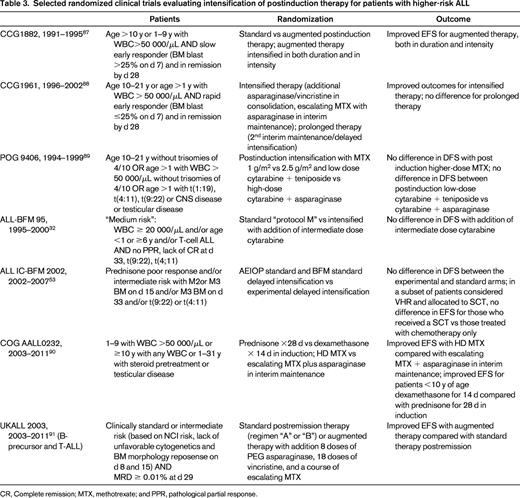
CR, Complete remission; MTX, methotrexate; and PPR, pathological partial response.
Improvements in outcomes for higher-risk patients in serial, nonrandomized trials have also been attributed to modifications of postinduction therapy, including the use of dexamethasone as the corticosteroid, the intensive use and formulation of asparaginase,4,56 and intensification of consolidation/reinduction treatment.32
Intensification of conventional chemotherapy has also been investigated within specific biological subsets of patients with ALL. Intrachromosomal amplification of chromosome 21 (iAMP) was found to be associated with a significantly inferior EFS in several cooperative group trials.45,61 Subsequent analysis identified that the poor outcome was driven primarily by patients with iAMP allocated to a standard-risk group and therefore treated with less intense therapies.62 Intensifying chemotherapy for this group of patients dramatically decreased the risk of relapse at 5 years from 70% to 16% over the course of 2 sequential trials.63
Similarly, patients with t(1:19) were found to have a significantly inferior outcome when treated with a regimen that was antimetabolite based,64 whereas when treated with intensified therapy, the outcome is significantly more favorable.65,66
Ongoing trials continue to evaluate the possibility of improving outcomes of children with high-risk ALL by intensifying postinduction chemotherapy, including the COG studies evaluating the impact of the addition of clofarabine for patients with very-high-risk precursor B-ALL and nelarabine for those with high-risk T-cell ALL. As opposed to improving outcomes with ongoing manipulation and intensification of conventional ALL therapies, identification of biologically driven subsets of ALL with unique druggable targets will likely lead to the next significant advances in ALL therapy. To date, this strategy has been shown to be highly effective for one subset of ALL, that of Ph+ ALL, which accounts for ∼3%-4% of cases. Favorable results for treatment of children with Ph+ ALL with the tyrosine kinase inhibitor imatinib in addition to intensive multiagent chemotherapy, leading to a 5-year disease-free survival (DFS) of 70%, has changed the standard of care for this group from that allocation to SCT in first remission (Figure 1).67-69

Data from COG protocol AALL0031. Shown are data with long-term follow-up of patients from COG protocol AALL0031 with Ph+ leukemia with no difference in 5-year DFS for patients in Cohort 5 (treated with continuous imatinib plus multiagent chemotherapy) compared with hematopoietic SCT in all cohorts, either from a related or unrelated donor. (Used with permission from Schultz et al, 2014.67 )
Data from COG protocol AALL0031. Shown are data with long-term follow-up of patients from COG protocol AALL0031 with Ph+ leukemia with no difference in 5-year DFS for patients in Cohort 5 (treated with continuous imatinib plus multiagent chemotherapy) compared with hematopoietic SCT in all cohorts, either from a related or unrelated donor. (Used with permission from Schultz et al, 2014.67 )
The identification of a subset of ALL patients who are Ph− but have a similar gene expression to those who are Ph+, called BCR-ABL1-like ALL, begs the question as to whether this group, that has been shown to have a worse prognosis with standard high-risk therapy,70 may benefit from targeted therapy, potentially with tyrosine kinase or JAK2 inhibitors.71 There are several published case reports describing children with ALL with induction failure found to have a BCR-ABL1-like disease who have subsequently had dramatic responses to tyrosine kinase inhibitors.72-74 Mechanisms for screening patients for BCR-ABL-like disease, evaluating for likelihood of response to tyrosine kinase inhibitors or JAK2 inhibitors in vitro, and considering the addition of targeted therapies to conventional chemotherapy are being planned within COG (Mignon Loh, personal communication).
Currently, there remains a population of children that can be identified as having an exceptionally high risk of having relapsed disease, for whom relatively poor outcomes with conventional therapy (including SCT) are expected. In the context of BFM therapy, patients with high MRD levels at the end of induction and consolidation had a 5-year DFS of 45% that was not improved by SCT.75 The 4-year OS for patients with hypodiploid ALL treated in a recent COG study with very intensive chemotherapy was 54%. With the limitations of small numbers, this study did not show an advantage of SCT as a component of therapy or improvement with intensified chemotherapy compared with historical controls.67 A retrospective review combining patients from multiple clinical trials showed a 32% 10-year OS for patients with initial induction failure.50 For those older than 6 years and those with T-cell ALL, there was a trend for improved outcome with SCT. A study of sibling donor allogeneic SCT in first remission versus chemotherapy for a group of patients defined as having very-high-risk disease, including those with induction failure, showed a DFS advantage with SCT.76 Lastly, despite intensified therapy, including high-dose cytarabine and methotrexate, infants with ALL with MLL translocations continue to have a poor outcome, with 5-year EFS between 30% and 40%.15 The use of SCT to intensify therapy for infants has not shown clear benefit, but the risk of relapse may be reduced for those in the highest risk group.16,77
TRM in high-risk ALL
Morbidity, both short-term and long-term, affects every patient treated for ALL. Despite improvements in supportive care, mortality as a consequence of therapy persists as a cause of death. A recent meta-analysis of randomized trials of newly diagnosed pediatric patients with ALL reported that the proportion of nonrelapse mortality was 3.6% overall and 1.4% in induction. In both periods, those classified as having high-risk leukemia had a significantly increased risk of nonrelapse mortality.78 Definitions of TRM in childhood ALL are variable and the majority of published clinical trials do not include descriptions of the definitions used.79 Variability in defining and reporting of TRM involves both inclusion and exclusion of deaths occurring immediately before the initiation of therapy or after completion of therapy, those that occur after SCT, and those from other causes not strictly related to disease or its therapy. The lack of definition of TRM makes comparison across trials difficult.
In the NOPHO ALL-92 and 2000 trials, 25% of all deaths on study were considered treatment related, with the majority being secondary to infection (72%), primarily bacterial infections, with bleeding or thrombosis, organ toxicity, or complications of tumor burden accounting for most of the remainder.80 Patients with high-risk ALL had a significantly higher risk of TRM than those with standard- or intermediate-risk disease (6.7% ± 0.9%, 1.7% ± 0.4%, and 2.4% ± 0.5%, respectively; p < .001). In the ALL IC-BFM 2002 trial, which expanded the access to trial participation to a large number of centers with relatively more limited resources, TRM accounted for 5% of deaths for those patients in complete remission, affecting 3% of those being treated for standard-risk disease and 13% of those being treated with high-risk regimens.53 Other factors that have been associated with TRM include age (<1 year or >15 years), female sex, SCT in first complete remission, and earlier treatment era.81,82 Patients with Down's syndrome and ALL constitute a unique group, with substantially higher TRM compared with non-Down's syndrome patients (7.7%±1% vs 2% ± <1% respectively, p < .0001) and the majority attributable to infection.25
The use of antibacterial prophylaxis during periods of prolonged neutropenia in adult patients with acute leukemia is supported by data from a meta-analysis showing its use to be associated with decreased risk of death,83 and is recommended in adult supportive care guidelines from both the National Comprehensive Cancer Network and the American Society of Clinical Oncology.84,85 Data on the use of antibacterial prophylaxis in children are extremely limited. Studies evaluating the utility of levofloxacin prophylaxis during ALL induction and in those being treated with intensive therapy for relapsed disease are being done through the Dana-Farber Cancer Institute consortium and COG, respectively.
Conclusion
The goals of therapy for every child newly diagnosed with ALL include maximizing the likelihood of cure while minimizing the risks of both acute and long-term side effects. Risk stratification, intensification of therapy for higher-risk patients, and, in the case of Ph+ ALL, adding targeted therapy have accounted for significant improvements in outlook for children and adolescents with high-risk disease. Despite progress, stratification schemes remain imperfect, with 1/3 of deaths in children with ALL in those who initially meet the criteria of favorable-risk disease.26 The next substantial steps in the efforts to improve the outcomes of children with ALL will likely include clinically accessible tools for measuring individual patient's disease features, with the potential for the identification of druggable targets. In addition, refining strategies for preventing TRM will allow for the tradition of remarkable progress in the care of children with ALL to continue.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Sarah Alexander, MD, The Hospital for Sick Children, 555 University Ave, Toronto, Ontario M5G 1X8, Canada; Phone: (416)813-7654, ext. 204068; Fax: (416)813-5327; e-mail: sarah.alexander@sickkids.ca.
