
Abstract
Iron is an ubiquitous metal of vital importance to the normal physiologic processes of many organisms. Over the last 2 decades, the discovery of mutations in genes leading to hereditary disorders of iron overload, iron deficiency, and iron maldistribution have accelerated our understanding of human iron homeostasis. This chapter provides an updated overview of the human iron cycle, regulation of iron homeostasis, and how perturbations in these homeostatic mechanisms lead to iron overload disease and provides strategies for the diagnosis of hereditary iron overload.
Learning Objectives
To appreciate the clinical and molecular modifiers of iron homeostasis
To develop an approach to the diagnosis of hereditary hemochromatosis
Introduction
Iron is an essential metal for many biological processes due to its ability to transfer electrons in reduction/oxidation reactions. This reactivity also provides the potential for great damage to biological systems if iron is not chaperoned through a tightly regulated network of iron-binding proteins and transporters. If the capacity of these iron-binding proteins is exceeded, “free” iron is capable of forming reactive oxygen species that may damage macromolecular cellular components such as nucleic acids, proteins, and lipids and lead to tissue damage. Iron homeostasis in humans is maintained almost exclusively at the level of intestinal absorption because evolution has not provided a physiologically regulated mechanism for iron excretion.
Iron homeostasis
Iron recycling and storage
The primary role of iron in mammals is to provide a binding site for oxygen in the heme moiety of hemoglobin. The average adult male has a total body iron content of ∼4 g, ∼2/3 of which is contained in the erythroid compartment. Under normal physiologic conditions, only 1-2 mg of the daily 20-25 mg of iron required to maintain erythropoiesis enters the body through carefully regulated intestinal absorption.1 Therefore, most iron required for the developing erythroid precursors in the BM is derived from the recycling of heme iron through the reticuloendothelial macrophage phagocytosis of senescent RBCs.
Most nonerythroid iron, 0.5–1 g in adult males, is stored in hepatocytes and macrophages and is easily mobilized for erythropoiesis. When iron stores are depleted through decreased dietary intake or intestinal absorption, increased requirements (eg, pregnancy) or loss (eg, intestinal blood loss), erythropoiesis becomes iron restricted and eventually results in the characteristic microcytic, hypochromic anemia of iron deficiency. In contrast, erythropoiesis in most iron overload states is relatively unperturbed; however, HFE hemochromatosis is associated with increases in hemoglobin, mean corpuscular volume, mean corpuscular hemoglobin, and mean corpuscular hemoglobin concentration that may be related to increased iron uptake and hemoglobin production in erythroid precursors.2 The excess iron absorbed in hereditary hemochromatosis is deposited in hepatocytes, cardiac myocytes, and multiple endocrine organs. This excess cellular iron deposition ultimately exceeds the capacity of iron-binding proteins, thereby leading to cellular damage, organ dysfunction, and eventually to clinical symptoms.
Ferritin is the primary intracellular iron storage protein, which forms multimeric complexes that facilitate iron sequestration and mobilization depending on cellular need. In contrast, hemosiderin is an amorphous and poorly bioavailable iron-containing conglomerate the presence of which is generally a pathologic sign of cellular iron excess. A soluble form of ferritin is also found in the plasma and is expelled primarily from reticuloendothelial macrophages and the liver; however, its biological role remains unclear. Serum ferritin correlates with iron stores in many conditions; however, caution must be taken before interpreting in the setting of inflammation and cellular injury.3
The large size and electrochemical properties of iron require that it be transported across the mammalian cell membranes by specific transmembrane proteins that mediate cellular iron uptake (import) and iron release (export). Although hepatocytes, intestinal mucosal cells, and macrophages possess specific carriers for iron import and export, erythroid cells only import iron and do not “release” iron until they are lysed or phagocytosed within the reticuloendothelial system.
Cellular iron import
Dietary nonheme iron, primarily in the ferric (Fe3+) state, is reduced to the ferrous (Fe2+) state in the acidic environment of the proximal duodenum by a brush border ferrireductase named Dcytb.4 Ferrous (Fe2+) iron is then cotransported with protons through the apical membrane of the duodenal enterocyte by divalent metal transporter 1 (DMT1).5 Some imported iron remains stored within the enterocyte cytoplasm as ferritin and the remainder is exported through the enterocyte basolateral membrane into the plasma via the transmembrane iron exporter ferroportin (FPN; described more fully in the Cellular iron export section).
Nonheme iron is imported into nonintestinal cell types from the plasma by the transferrin (TF) cycle (for review, see Chen and Paw6 ). Almost all plasma iron exists bound to the abundant glycoprotein TF. Each TF molecule binds 2 Fe3+ iron atoms with high affinity, thereby maintaining its solubility and nonreactivity and allowing it to circulate in a safe form. The TF cycle begins with iron-loaded plasma TF binding with high affinity to TF receptor 1 (TFR1) on the cell surface and being endocytosed. The endosome is then acidified, prompting the release of iron from TF and the empty TF and TFR1 return to the cell surface and are available to repeat the cycle again. The unbound Fe3+ iron in the acidified endosome is reduced and transported out of the endosome into the cytoplasm by DMT1.
Despite significant advances in our understanding of absorption and regulatory mechanisms of nonheme iron, the mediators of heme iron import, particularly during intestinal absorption, remains enigmatic (for review, see Yuan et al7 and Hamza and Korolnek8 ). It is unclear whether intestinal heme absorption uses membrane-bound transporters or endocytic uptake.8 The only membrane-bound heme importer characterized is the heme-responsive gene-1, (HRG-1), initially identified by homology to heme transporters found in Caenorhabditis elegans.9,10 HRG-1 localizes to the phagolysosomes of macrophages,9,11 suggesting a role in the import of heme during the recycling of senescent erythrocytes. Most intracellular heme will have its iron liberated by hemoxygenase, allowing storage in ferritin or export via FPN. However, cellular heme export has been postulated as a method of cellular detoxification and transfer of heme between cells.12 Feline Leukemia Virus sub-group C Receptor 1a (FLVCR1a), ATP-binding cassette transporter sub-family G member 2 (ABCG2),12 and, most recently, the multidrug resistance protein (MRP-5)13 have been implicated as heme exporters.
Cellular iron export
Some imported iron remains stored intracellularly as ferritin and the remainder is exported. The only known mammalian iron exporter is FPN,14,15 which is expressed at all sites involved in cellular Fe2+ iron export to the plasma, including the basolateral membranes of duodenal enterocyte, macrophages, hepatocytes, and in the placenta.16
Cellular iron export is dependent on the copper-containing ferroxidases ceruloplasmin and hephaestin.17 These enzymes oxidize exported Fe2+ iron, returning it to its Fe3+ state and thus promoting its binding to plasma TF. Deficiencies of these ferroxidases appear to result in impaired iron export from the cells/tissue where they are expressed and appear to result in cellular iron accumulation.16
Regulation of iron homeostasis
Iron homeostasis requires carefully coordinated regulation of intestinal iron absorption, cellular iron import/export, and iron storage. Because humans have no physiological mechanism for iron excretion, the control of intestinal iron absorption is the primary mechanism for determining overall iron balance. A small fraction of dietary iron is imported into the enterocyte and only a fraction is exported from the enterocyte into the plasma. Iron remaining within enterocytes is lost from the body through their physiologic sloughing into the gut lumen.
Intestinal iron absorption is regulated primarily through the expression of hepcidin, a circulating 25-aa hormone synthesized by the liver and filtered into the urine.18 Hepcidin acts as a negative regulator of cellular iron export from cells, thus limiting iron availability to the plasma. As a negative regulator, decreased hepcidin leads to enhanced intestinal absorption and macrophage iron release, elevated plasma iron levels, and iron loading.
Hepcidin exerts its central role in iron homeostasis through its effect on FPN, the only known receptor for hepcidin and cellular iron exporter. Hepcidin binds to FPN at the cell surface, resulting in endocytosis and lysosomal degradation,19 blocking of iron efflux into the plasma, hypoferremia, and iron-restricted erythropoiesis.
Clinically, plasma hepcidin concentration is modulated by several “regulators.”20 Hepcidin is increased by increased iron stores or inflammation and is decreased by hypoxia, increased erythropoietic activity, or testosterone.21 In the setting of conflicting influences on hepcidin expression, the negative regulation by erythropoietic activity is dominant (eg, non-transfusion-dependent thalassemia). The physiologic regulation of hepcidin expression is controlled at the level of transcription by a complex network resulting from the interaction of multiple proteins and cascades (Figure 1). Although a great deal remains to be elucidated regarding the molecular control of hepcidin expression, much has been learned through studies of human diseases or mouse models in which hepcidin is dysregulated.22

Regulators of hepcidin expression. Hepcidin is a common effector of 4 known regulators of iron homeostasis. Iron stores, erythropoietic demand, hypoxia, and inflammation all act by modulating hepatocyte production of hepcidin. Increased iron stores and inflammation both appear to increase hepcidin expression, primarily through signal transduction via the SMAD and JAK/STAT pathways, respectively. Increased/ineffective erythropoietic drive and hypoxia appear to decrease hepcidin expression, although the mechanisms of their control of hepcidin expression remain to be fully elucidated.
Regulators of hepcidin expression. Hepcidin is a common effector of 4 known regulators of iron homeostasis. Iron stores, erythropoietic demand, hypoxia, and inflammation all act by modulating hepatocyte production of hepcidin. Increased iron stores and inflammation both appear to increase hepcidin expression, primarily through signal transduction via the SMAD and JAK/STAT pathways, respectively. Increased/ineffective erythropoietic drive and hypoxia appear to decrease hepcidin expression, although the mechanisms of their control of hepcidin expression remain to be fully elucidated.
Hepcidin regulation by plasma iron and tissue iron stores is mediated through the bone morphogenic protein (BMP) in the sons of mothers against decapentaplegic (SMAD) pathway. In the iron-replete state, BMP6 binds to its receptor (BMPR) and their coreceptor hemojuvelin (HJV), which triggers a SMAD-dependent signaling cascade to increase hepcidin transcription in hepatocytes. The sensor for iron repletion is not nearly as clear, but appears to involve interactions among the BMPR/HJV complex, HFE, and TF receptor 2 (TFR2).16 Positive flux through this BMP/SMAD pathway can be dampened by the cleavage of HJV by the membrane-associated serine protease matriptase-2 encoded by TMPRSS6.22
Hepcidin up-regulation by inflammation appears independent of BMP6 and HJV and mediated by inflammatory cytokines, especially IL-6, which binds to its receptor, activating JAK2 signaling and STAT3 phosphorylation.23 The inflammatory pathway requires the integrity of the BMP-SMAD pathway (particularly SMAD4) to fully activate hepcidin.24 The inflammatory cytokine Activin B also induces SMAD signaling and acts synergistically with IL-6 JAK2/STAT-3 signaling to up-regulate hepcidin expression.25
Hepcidin is clearly down-regulated in the setting of hypoxia, anemia, and increased/ineffective erythropoietic activity; however, the identification of the mediator(s) and molecular mechanism remains unknown. The most commonly held hypothesis is that the BM produces a hepcidin-suppressing mediator in response to physiologic erythropoietin or pathophysiologic ineffective erythropoiesis. Growth differentiation factor 15 (GDF15) and twisted-gastrulation 1 (TWSG1) are BMP family members postulated to be suppressors of hepcidin in thalassemia26,27 ; however, their contribution remains uncertain.28 Erythroferrone (ERFE) has recently been described as a protein that mediates hepcidin suppression during stress erythropoiesis and ineffective erythropoiesis.29
Dysregulation of hepcidin expression results in several hereditary and acquired disease entities. Inappropriately elevated levels of hepcidin can result in iron-restricted anemia that is acquired (eg, anemia of chronic inflammation) or more rarely hereditary (eg, iron-refractory iron deficiency anemia). Conversely, inappropriately low levels of hepcidin result in iron overload that can be hereditary (eg, hereditary hemochromatosis) or secondary (eg, anemias with ineffective erythropoiesis). The remainder of this review will concentrate on hereditary disorders of iron overload.
Iron overload states
Although the earth is an iron-rich environment, acquired iron deficiency remains a global public health concern, with an estimated 1 billion persons affected. In contrast, most hereditary defects of iron metabolism result in iron overload rather than iron deficiency. The known genetic defects in many hereditary disorders of iron overload result in the aberrant regulation of iron absorption and storage. Clinically significant hereditary iron overload is almost exclusively an adult phenomenon, but there are rare pediatric forms. A summary of these disorders and their genetics can be found in Table 1.
Hereditary iron overload disorders
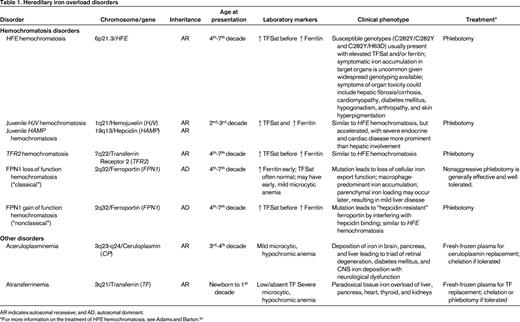
AR indicates autosomal recessive; and AD, autosomal dominant.
*For more information on the treatment of HFE hemochromatosis, see Adams and Barton.61
HFE hemochromatosis
Originally recognized in the mid-19th century, this form of hereditary hemochromatosis was not linked to mutations in the HFE gene until 1996.30 HFE encodes atypical HLA class I-like membrane associated protein with a similar structure to other class I HLA membrane-associated proteins, but has no known function in antigen presentation. HFE is widely expressed and is known to bind with TFR131 to decrease its affinity for iron-loaded TF.32 Although the role of HFE in the regulation of hepcidin transcription remains unclear, patients with HFE hemochromatosis have inappropriately low hepcidin expression,33 which explains their increased intestinal iron absorption and increased reticuloendothelial iron release into the plasma.
Although HFE hemochromatosis has a worldwide distribution, it is the most common autosomal recessive disorder of northern European heritage, apparently having descended from a common Celtic ancestor.34 Most HFE hemochromatosis patients are homozygous for a p.Cys282Tyr (C282Y) amino acid substitution. The estimated prevalence of the C282Y allele in Caucasian Americans is 10%–15%, consistent with a homozygote prevalence estimated at 1 in every 200-250 persons.35-37
Some clinical laboratories commonly report other HFE mutations. The p.His63Asp (H63D) amino acid substitution is occurs in ∼20% of the global population.36,38 The p.Ser65Cys (S65C) amino acid substitution is also reported by some, but the clinical relevance is not clear. Other very rare (“private”) HFE mutations can be found in trans to the C282Y and can explain some cases of rapid iron overload out of keeping with the C282Y carrier status.39 HFE sequencing (beyond genotyping for C282Y and H63D) may be difficult to obtain clinically and reimbursement by insurance payer may also be challenging.
Despite the relatively high prevalence of the HFE mutations in some populations, the penetrance of HFE hemochromatosis is low. Although C282Y homozygotes may develop biochemical evidence of significant iron loading in adolescence (an elevation in TF saturation usually precedes elevation of ferritin), usually only a small proportion ever develop sufficient iron overload to cause symptomatic clinical manifestations related to tissue or organ damage.40 Depending on the diagnostic criteria used, clinically significant iron overload is observed in only 1%–75% of C282Y homozygotes,37,41 with a recent European meta-analysis showing an overall penetrance of 13.5%.42
The pathophysiologic consequences of the H63D mutation, and particularly the implications for C282Y/H63D compound heterozygotes, remain somewhat less certain. Only a small fraction of C282Y/H63D heterozygotes appear to develop biochemical evidence of iron loading and generally do not develop overt iron overload43 and clinicians should consider the contribution of other factors leading to iron overload in patients of this genotype.
HFE hemochromatosis was classically described as the triad of cirrhosis, diabetes mellitus, and skin hyperpigmentation developing in the in the 4th-7th decade of life and thus earning the description “bronze diabetes.” However, with the availability of genetic diagnosis since the late 1990s, the clinical presentation of HFE hemochromatosis has changed as a result of earlier diagnosis due to routine chemistry screening and screening relatives of affected individuals. Evaluations of large populations of in C282Y homozygotes or C282Y/H63D compound heterozygotes has revealed that the frequency of classically related nonspecific symptoms (eg, fatigue, arthralgias, affective disorders) and symptoms of end organ damage (eg, cardiomyopathy, arrhythmias, diabetes mellitus, sexual dysfunction) are uncommon,41 and in one study they did not differ significantly from wild-type controls.37 Men are significantly more likely to have symptomatic iron overload than women41 and, although this has been attributed to physiologic blood loss of menstruation and pregnancy or sex-related disease modifier genes,44 it may be more related to the testosterone-mediated suppression of hepcidin in males.21
Juvenile hemochromatosis (HJV hemochromatosis and HAMP hemochromatosis)
Juvenile hemochromatosis is a rare, autosomal-recessive disorder with a phenotype of accelerated iron loading that usually presents before or during the 2nd decade of life. Genetic linkage analysis of multiple affected families revealed 2 distinct genetic entities with a common phenotype. Homozygous or compound heterozygous mutations in HJV or hepcidin (HAMP)45 result in the juvenile hemochromatosis phenotype. Both genotypes appear to have high penetrance, but HJV mutations are responsible for a majority of reported juvenile hemochromatosis cases.46
As discussed previously, hemojuvelin is a critical BMPR coreceptor critical for SMAD-dependent signaling and hepcidin transcription. Mutations in HJV or hepcidin itself results in severely decreased hepcidin transcription and plasma levels with increased intestinal iron absorption and reticuloendothelial iron release, leading to rapid iron accumulation in target organs.
In contrast to HFE hemochromatosis, patients with juvenile hemochromatosis are more likely to present with endocrine failure (hypogonadism) or cardiomyopathy and hepatic disease is less prominent.47 Affected patients usually die from cardiac failure by age 30 years if untreated.48 The most effective current treatment is aggressive serial phlebotomy.
TFR2 hemochromatosis
TFR2 shares significant sequence homology with TFR1 and, although it can bind TF, it does so with lower affinity.49 The normal physiologic role of TFR2 in iron metabolism is not yet well understood, but appears to be involved as the “sensor” of iron status that regulates hepcidin expression.16,22 The prevalence of TFR2 mutations is low relative to HFE mutations, but they appear to have a more global ethnic distribution.46 One might expect that the loss of TFR2 function would result in decreased cellular iron uptake, but mutations in TFR2 are associated with an autosomal-recessive, adult-onset iron loading phenotype very similar to HFE hemochromatosis, whichi s associated with a predilection for hepatocellular iron deposition.50 The most effective current treatment is also serial phlebotomy.
FPN1 hemochromatosis
FPN1 hemochromatosis appears to be the most common form of non-HFE hemochromatosis.51 It is distinguished from other iron overload disorders by its autosomal-dominant inheritance and 2 phenotypic subtypes depending on the functional consequences of the pathological mutations in FPN1, the only cellular iron exporter.
Loss-of-function FPN1 hemochromatosis (“classical”) is associated with impaired iron release from macrophages, with reticuloendothelial accumulation of iron and iron-restricted erythropoiesis resulting in a mild, hypochromic anemia. Serum ferritin levels rise early and out of proportion to TF saturation, a pattern in contrast to the early elevation in TF saturation seen in HFE hemochromatosis. Although FPN1 hemochromatosis is generally milder than HFE hemochromatosis, the complete spectrum of iron overload morbidity has been demonstrated. Phlebotomy is a generally effective therapy; however, given the frequent presence of mild anemia and sequestration of iron within macrophages, patients usually require a gentler iron depletion program.52
Rarer cases of gain-of-function FPN1 hemochromatosis (“nonclassical”) are related to mutations in FPN1 that lead to impaired hepcidin binding (“resistance”) and constitutive iron export from enterocytes and macrophages.51 Gain-of-function FPN1 hemochromatosis mutations lead to elevated TF saturation and hepatocellular/parenchymal iron overload that is phenotypically very similar to HFE hemochromatosis.
Aceruloplasminemia
Ceruloplasmin is a plasma copper-containing protein synthesized primarily in the liver. It is also expressed in a glycosylphosphatidylinositol-linked isoform on glial cells of the CNS.53 Ceruloplasmin is a ferrioxidase that has a vital function to iron mobilization from the tissues by converting Fe2+ to Fe3+.
Aceruloplasminemia is an extremely rare, autosomal-recessive disorder caused by missense/nonsense mutations in the ceruloplasmin gene (CP) found on chromosome 3q21.54 The absence of ferrioxidase ceruloplasmin does not affect copper homeostasis, but impairs mobilization of recycled iron stores from the reticuloendothelial system, resulting in low TF saturation, iron-restricted anemia, and visceral iron retention similar to gain-of-function FPN1 hemochromatosis. However, aceruloplasminemia can be clinically distinguished from FPN disease and the more common HFE hemochromatosis by the unique triad of early retinal degeneration, diabetes mellitus, and CNS iron deposition with neurological dysfunction. Onset of retinal degeneration and diabetes mellitus may occur as early as the 3rd decade, followed by neurological symptoms as much as a decade later. Neurological symptoms, including ataxia, involuntary movements, and cognitive dysfunction, are likely related to direct neurotoxicity of iron deposition in the CNS.55 Treatment with iron chelators, specifically improving the hepatic iron overload and diabetes, has met with some success, but is ineffective for mobilization of brain iron and neurological symptoms. In addition, chelation may be limited by accentuation of the associated iron-restricted anemia.54
Atransferrinemia
Congenital deficiency of TF, the primary plasma iron-binding protein, is another extremely rare autosomal-recessive disorder due to mutations of the TF gene.56 Without plasma TF to deliver iron to erythroid precursors, affected individuals develop a severe, microcytic, hypochromic, iron-deficiency anemia phenotype requiring erythrocyte transfusions from birth.57 However, this iron-restricted state leads to enhanced intestinal absorption of iron that is ineffectively transported in the plasma and imported into erythroid precursors. The resulting non-TF-bound iron is avidly imported by parenchymal cells by an unclear mechanism and leads to a paradoxical iron overload of nonerythropoietic tissues. Tissues affected are similar to those seen in hemochromatosis, including liver, pancreas, heart, thyroid, and kidneys. Reticuloendothelial macrophages may be spared until the development of transfusion-related hemosiderosis. Serum ferritin is elevated and serum iron and total iron-binding capacity are very low. Supportive care with erythrocyte transfusions may relieve the anemia. Recurrent infusions of fresh-frozen plasma containing TF combined with chelation therapy or phlebotomy may be effective.56
Diagnostic approach to hereditary iron overload
The diagnosis of hereditary iron overload relies on the careful evaluation of clinical phenotype, family history, and laboratory assessment. The rational use of a genetic testing algorithm will prevent the excessive use of resources (Figure 2).43,58

Guide to the evaluation of hereditary iron overload. After ruling out acquired causes of hyperferritinemia, TF saturation and clinical phenotype directs further rational genetic evaluation of hereditary iron overload. See text for more details. MAS indicates macrophage activation syndrome; HLH, hemophagocytic lymphohistiocytosis; MDS, myelodysplastic syndrome; LIC, liver iron concentration; and HH, hereditary hemochromatosis.
Guide to the evaluation of hereditary iron overload. After ruling out acquired causes of hyperferritinemia, TF saturation and clinical phenotype directs further rational genetic evaluation of hereditary iron overload. See text for more details. MAS indicates macrophage activation syndrome; HLH, hemophagocytic lymphohistiocytosis; MDS, myelodysplastic syndrome; LIC, liver iron concentration; and HH, hereditary hemochromatosis.
Large screening studies in HFE hemochromatosis have revealed that only ∼70% of adult C282Y homozygotes have elevated ferritin, and only a very small fraction of these have a symptomatic iron overload phenotype.36,37,41 Although uncommon, the clinician should still be able to recognize the symptomatic iron overload phenotype (hepatic or cardiac dysfunction, early onset atypical arthropathy, diabetes, sexual dysfunction, affective disorders, or hyperpigmentation).40 The clinician must also rule out secondary causes of iron overload (eg, chronic transfusion therapy, ineffective erythropoiesis, chronic supplemental iron) and alternative causes for elevated ferritin and modification of iron burden (eg, metabolic syndrome, inflammatory states, cell damage).
Family history suggestive of an inherited disease is also very important. Specific questioning regarding iron or liver abnormalities, race/ethnicity, and possible consanguinity are required. If a family history is positive, the clinician should attempt to determine the mode of inheritance (eg, recessive/dominant).
Laboratory documentation of increased iron stores is often the first step in screening/diagnosing hereditary hemochromatosis and include TF saturation (TFSat), unsaturated iron-binding capacity, and serum ferritin. TFSat is the ratio of serum iron to total iron-binding capacity expressed as a percentage. Previous recommendations to obtain fasting samples for TfSat have been shown to be unnecessary because fasting does not affect the sensitivity or specificity of detecting HFE hemochromatosis.59 Unsaturated iron-binding capacity has been shown to be a modestly better predictor of HFE hemochromatosis than TFSat, with the added advantage of lower cost and being a single direct measurement rather than a ratio60 ; however, the most recent American Association for the Study of Liver Diseases (AASLD) practice guideline uses TFSat in its diagnostic algorithm. Persistently elevated early morning fasting TFSat >45% is generally considered diagnostic of iron overload.40
Serum ferritin provides a useful assessment body iron stores and confirmatory evidence of iron overload in the setting of an elevated TFSat. However, serum ferritin can be falsely elevated in some clinical settings such as inflammation, hepatic injury, and rapid cell turnover. Therefore, interpretation of an elevated ferritin must made in the broader clinical context. In Caucasians, an elevated TFSat should prompt assessment of the common HFE genotypes. Discovery of C282Y homozygosity confirms HFE hemochromatosis. Compound heterozygotes for HFE C282Y/H63D generally do not develop overt iron overload and clinicians should consider the contribution of other factors leading to iron overload. If the HFE genotyping is negative, one must consider rare private HFE mutations with complete HFE sequencing and non-HFE form of hereditary hemochromatosis. Non-HFE hemochromatosis etiologies can generally be divided into those with rapid loading and “juvenile” onset (<30 years of age) and others mimicking the natural history of HFE hemochromatosis. Younger patients should have sequencing of HJV and HAMP to assess for these forms of juvenile hemochromatosis, respectively. Older patients should have sequencing of TFR2 to assess for TFR2 hemochromatosis and FPN1 sequencing for mutations leading to FPN “resistance” and loss-of-function FPN1 hemochromatosis type 4B.
Much less commonly, a patient without acquired causes will present with isolated hyperferritinemia and a normal TF saturation. Confirmation of increased liver iron content by MRI or liver biopsy should prompt further careful evaluation of the hematologic and neurologic phenotype. Predominant macrophage iron loading on liver biopsy should prompt FPN1 sequencing for loss-of-function FPN1 hemochromatosis. The presence of microcytic, hypochromic anemia should prompt measurement of serum TF for consideration of hereditary hypo/atransferrinemia. If there are neurological symptoms in addition to anemia, ceruloplasmin should be measured in consideration of hereditary aceruloplasminemia. If isolated hyperferritinemia is discovered without an elevated liver iron concentration but with the presence of cataracts or brain iron loading, consideration should be given to sequencing ferritin light chain (FTL) for hyperferritinemia-cataract syndrome. Hyperferritinemia-cataract syndrome is more common than hereditary hypo/atransferrinemia and, although it is autosomal recessive, it can mimic the autosomal-dominant FPN1 loss-of-function hemochromatosis with elevated ferritin and normal TFSat.
Occasionally, a patient presents with what appears to be a hereditary hemochromatosis that does not fall neatly into the algorithm presented in Figure 2. Some of these individuals may harbor mutations in an unknown gene related to iron metabolism or hepcidin expression and others may have complex genotypes with single mutated/polymorphic alleles of 2 or more of the known iron homeostasis genes presented.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Matthew M. Heeney, MD, Boston Children's Hospital, 300 Longwood Ave., Fegan 702, Boston, MA 02115; Phone: (617)919-3242; Fax: (617)730-0641; e-mail: matthew.heeney@childrens.harvard.edu.
