
Abstract
Mastocytosis is a rare disease characterized by KIT-driven expansion and accumulation of neoplastic mast cells in various tissues. Although mediator symptoms related to mast cell activation can impose a symptom burden in cutaneous disease and across the spectrum of systemic mastocytosis subtypes, the presence of an associated hematologic neoplasm and/or organ damage denotes advanced disease and the potential for increased morbidity and mortality. In addition to the revised 2016 World Health Organization classification of mastocytosis, a new diagnostic and treatment toolkit, tethered to enhanced molecular characterization and monitoring, is poised to transform the management of patients with advanced systemic mastocytosis (advSM). Although the efficacy of midostaurin and novel selective KIT D816V inhibitors, such as avapritinib (BLU-285), have validated KIT as a therapeutic target, the clinical and biologic heterogeneity of advSM requires that we reimagine the blueprint for tackling these diseases and use tools that move beyond KIT-centric approaches.
Learning Objectives
Review the revised 2016 World Health Organization classification of mastocytosis and diagnostic pearls related to the workup of advanced systemic mastocytosis (advSM)
Understand the role of KIT D816V and other myeloid mutation profiling in the diagnosis, prognostication, and treatment monitoring of advSM
Identify the role of midostaurin and novel KIT inhibitors in the treatment of advSM
Patient scenario
A 61-year-old man with JAK2 V617F mutation–positive, Dynamic International Prognostic Scoring System-Plus Intermediate-2–risk primary myelofibrosis (PMF) presented with fatigue, night sweats, symptomatic splenomegaly 12 cm below the left costal margin, and a 7-kg weight loss. After 18 months of sustained improvement in symptoms and splenomegaly on ruxolitinib, he develops regrowth of splenomegaly, new hepatomegaly with elevation of the serum alkaline phosphatase level to 340 IU/L, and paracentesis-dependent ascites. A complete blood count reveals a white blood cell count of 13 × 109/L; over the last 2 months, the hemoglobin has decreased from 10.6 to 9.3 g/dL, and the platelet count has decreased from 115 to 74 × 109/L. The differential reveals mild myeloid immaturity and leukoerythroblastosis. A bone marrow (BM) aspirate is a dry tap; the core biopsy is hypercellular with marked reticulin fibrosis and atypical megakaryocyte clustering without increased blasts. However, a few multifocal aggregates of spindle-shaped cells are noted. Immunohistochemistry (IHC) with CD117, tryptase, and CD25 highlights abnormal mast cells (MCs) comprising 10% of the marrow cellularity. Chromosome analysis is normal. The marrow findings prompt additional diagnostic testing: a serum tryptase level is 220 ng/mL (normal <11.4) and KIT D816V allele–specific polymerase chain reaction (PCR) on the peripheral blood is positive. Myeloid mutation panel testing confirms KIT D816V and JAK2 V617F (variant allele frequencies [VAFs] of 38% and 60%, respectively) as well as pathogenic ASXL1 and RUNX1 mutations. A liver biopsy is discussed with the patient.
Introduction
Mastocytosis encompasses a spectrum of disorders characterized by abnormal expansion and accumulation of neoplastic MCs in different organs, including the skin, BM, lymph nodes, spleen, liver, and gastrointestinal tract. Normal MCs play an important role in the regulation of immunoglobulin E (IgE)–mediated allergic responses, inflammation, and the innate and adaptive immune responses to infection.1 Abnormal activation and accumulation of MCs can lead to mediator symptoms and organ damage. Several recent developments in the field of MC neoplasms include an updated classification, extended molecular profiling beyond KIT D816V to improve prognostication, and new consensus response criteria for advanced systemic mastocytosis (advSM). These new tools together with the approval of midostaurin and the emergence of selective KIT D816V inhibitors have created a unique opportunity to impact the natural history of these poor-prognosis neoplasms.
Classification
In the revised 2016 World Health Organization (WHO) classification of hematopoietic and lymphoid tumors, mastocytosis was removed as a subtype of myeloproliferative neoplasms (MPNs) and designated as a separate major disease category.2 The mastocytosis classification is broadly divided into cutaneous mastocytosis, systemic mastocytosis (SM), and MC sarcoma; due to its extreme rarity, extracutaneous mastocytoma was eliminated as a disease entity.2,3 Although the 2016 diagnostic criteria for SM remain largely unchanged (Table 1),2 a few modifications were made to its subtypes. (1) Smoldering systemic mastocytosis (SSM) was removed as a subtype of indolent systemic mastocytosis (ISM)2 owing to its increased risk of progression to more advanced disease and lower overall survival (OS) compared with ISM, which has a life expectancy similar to age-matched healthy controls.4,5 (2) A nomenclature revision permits the simpler term systemic mastocytosis with an associated hematologic neoplasm (SM-AHN) to be used interchangeably with SM with an associated hematologic non-MC lineage disease (SM-AHNMD),2 which will likely be phased out over time. B findings (indicating a high burden of MCs without evidence of organ damage) and C findings (organ damage produced by neoplastic MC infiltration) are used to discriminate SSM (≥2 B findings) from aggressive systemic mastocytosis (ASM; ≥1 C finding[s]) (Table 1).2 ASM in transformation is a new variant delineated by 5% to 19% MCs on the BM aspirate and reflects accelerated disease.6 Mast cell leukemia (MCL) is defined histopathologically by the presence of ≥20% MCs on a BM aspirate smear.2 In some cases, discordance may exist between the high burden of MCs identified on a core biopsy and a smaller than expected percentage of MCs on an aspirate, because such cells may be encased within fibrotic aggregates, producing a paucicellular specimen or dry tap. MCL has been divided into a chronic form without obvious organ damage and a more aggressive variant, termed acute MCL, where organ damage (C findings) is present.2,6 advSM is an umbrella term used to refer to SM-AHN, ASM, and MCL, because these patients are usually afflicted by organ damage and exhibit reduced OS.
The 2016 WHO diagnostic criteria for SM, SM variants, and B and C findings
| Diagnostic criteria |
|---|
| SM diagnostic criteria* |
| Major |
| Multifocal, dense infiltrates of MCs (15 or more in aggregates) detected in sections of BM and/or another extracutaneous organ |
| Minor |
| In biopsy sections of BM or other extracutaneous organs, >25% of the MCs in the infiltrate are spindle shaped or have atypical morphology, or of all MCs in BM aspirate smears, >25% are immature or atypical MCs |
| Detection of an activating point mutation at codon 816 of KIT in BM, blood, or another extracutaneous organ |
| MCs in the BM, blood, or another extracutaneous organ express CD25, with or without CD2, in addition to normal MC markers |
| Serum total tryptase persistently >20 ng/mL in the absence of an associated clonal myeloid disorder |
| Variants of SM |
| Indolent SM† |
| No C findings |
| No evidence of an associated hematologic neoplasm |
| Low MC burden |
| Skin lesions are often present |
| Smoldering SM |
| ≥2 B findings; no C findings |
| No evidence of an associated hematologic neoplasm |
| Does not meet criteria for MC leukemia |
| SM with an associated hematologic neoplasm‡ |
| Meets the general criteria for SM as well as associated hematologic neoplasms classified as a distinct entity in the WHO classification |
| Aggressive SM§ |
| ≥1 C finding |
| Does not meet criteria for MC leukemia |
| MC leukemia‖ |
| BM aspirate smears show >20% MCs |
| BM biopsy shows diffuse infiltration (usually dense) by atypical, immature MCs |
| B and C findings |
| B findings indicate a high burden of MCs and expansion of the neoplastic process into multiple hematopoietic lineages without evidence of organ damage |
| High MC burden (shown on BM biopsy): >30% infiltration of cellularity by MCs and serum total tryptase >200 ng/mL |
| Signs of dysplasia or myeloproliferation in non-MC lineage(s), but criteria are not met for definitive diagnosis of an associated hematologic neoplasm, with normal or only slightly abnormal blood counts |
| Hepatomegaly without impairment of liver function, palpable splenomegaly without hypersplenism, and/or lymphadenopathy on palpation or imaging |
| C findings are indicative of organ damage produced by MC infiltration (should be confirmed by biopsy if possible) |
| BM dysfunction caused by neoplastic MC infiltration manifested by ≥1 cytopenia: absolute neutrophil count <1.0 × 109/L, hemoglobin level <10 g/dL, and/or platelet count <100 × 109/L |
| Palpable hepatomegaly with impairment of liver function, ascites, and/or portal hypertension |
| Skeletal involvement, with large osteolytic lesions with or without pathological fractures (pathological fractures caused by osteoporosis do not qualify as a C finding) |
| Palpable splenomegaly with hypersplenism |
| Malabsorption with weight loss due to gastrointestinal MC infiltrates |
| Diagnostic criteria |
|---|
| SM diagnostic criteria* |
| Major |
| Multifocal, dense infiltrates of MCs (15 or more in aggregates) detected in sections of BM and/or another extracutaneous organ |
| Minor |
| In biopsy sections of BM or other extracutaneous organs, >25% of the MCs in the infiltrate are spindle shaped or have atypical morphology, or of all MCs in BM aspirate smears, >25% are immature or atypical MCs |
| Detection of an activating point mutation at codon 816 of KIT in BM, blood, or another extracutaneous organ |
| MCs in the BM, blood, or another extracutaneous organ express CD25, with or without CD2, in addition to normal MC markers |
| Serum total tryptase persistently >20 ng/mL in the absence of an associated clonal myeloid disorder |
| Variants of SM |
| Indolent SM† |
| No C findings |
| No evidence of an associated hematologic neoplasm |
| Low MC burden |
| Skin lesions are often present |
| Smoldering SM |
| ≥2 B findings; no C findings |
| No evidence of an associated hematologic neoplasm |
| Does not meet criteria for MC leukemia |
| SM with an associated hematologic neoplasm‡ |
| Meets the general criteria for SM as well as associated hematologic neoplasms classified as a distinct entity in the WHO classification |
| Aggressive SM§ |
| ≥1 C finding |
| Does not meet criteria for MC leukemia |
| MC leukemia‖ |
| BM aspirate smears show >20% MCs |
| BM biopsy shows diffuse infiltration (usually dense) by atypical, immature MCs |
| B and C findings |
| B findings indicate a high burden of MCs and expansion of the neoplastic process into multiple hematopoietic lineages without evidence of organ damage |
| High MC burden (shown on BM biopsy): >30% infiltration of cellularity by MCs and serum total tryptase >200 ng/mL |
| Signs of dysplasia or myeloproliferation in non-MC lineage(s), but criteria are not met for definitive diagnosis of an associated hematologic neoplasm, with normal or only slightly abnormal blood counts |
| Hepatomegaly without impairment of liver function, palpable splenomegaly without hypersplenism, and/or lymphadenopathy on palpation or imaging |
| C findings are indicative of organ damage produced by MC infiltration (should be confirmed by biopsy if possible) |
| BM dysfunction caused by neoplastic MC infiltration manifested by ≥1 cytopenia: absolute neutrophil count <1.0 × 109/L, hemoglobin level <10 g/dL, and/or platelet count <100 × 109/L |
| Palpable hepatomegaly with impairment of liver function, ascites, and/or portal hypertension |
| Skeletal involvement, with large osteolytic lesions with or without pathological fractures (pathological fractures caused by osteoporosis do not qualify as a C finding) |
| Palpable splenomegaly with hypersplenism |
| Malabsorption with weight loss due to gastrointestinal MC infiltrates |
A diagnosis of SM requires 1 major plus 1 minor criterion or ≥3 minor criteria.
BM mastocytosis is a subtype of indolent SM characterized by BM involvement and no skin lesions.
Usually, a myeloid neoplasm (ie, MDS, MPN, MDS/MPN, chronic eosinophilic leukemia, not otherwise specified, or acute myeloid leukemia). Lymphoid neoplasms (ie, multiple myeloma and chronic lymphocytic leukemia) constitute <10% of associated hematologic neoplasms.
Aggressive SM in transformation is a subvariant characterized by 5% to 19% MCs on BM aspirate smears and reflects evolution toward MC leukemia.
The aleukemic MC leukemia variant (in which MCs account for <10% of circulating white blood cells) is more common than cases with ≥10% circulating MCs, which are more aggressive; MC leukemia has also been divided into chronic (C findings absent) and acute (C findings present).
The diagnostic toolkit
Due to its protean manifestations, a high index of suspicion is needed to make the diagnosis of mastocytosis. Patients commonly experience diagnostic delay and may undergo consultation with a cadre of subspecialists, including allergists/immunologists, gastroenterologists, dermatologists, and hematologists/oncologists. Mastocytosis should be suspected in patients presenting with skin involvement (ie, urticaria pigmentosa/maculopapular cutaneous mastocytosis), recurrent MC activation (mediator) symptoms (ie, flushing, diarrhea, and anaphylaxis), an increased serum tryptase level, and/or the presence of B or C findings.7 Most adults with cutaneous disease also exhibit systemic involvement.8,9
Histopathology
Regardless of whether patients present with indolent or advanced forms of SM, evaluation of a biopsy from the BM or other extracutaneous tissue(s) by an experienced pathologist (and/or center with clinical MC expertise) is a critical component of the diagnostic toolkit (Figure 1). IHC of the affected tissue for CD117 (KIT), CD25, and tryptase is important for the identification of atypical MC populations.2 Flow cytometry using CD117, CD25, and CD2 is a complementary tool in the diagnosis of SM; flow cytometric characterizations of MCs are rare event analyses, and optimal techniques for enumeration of MCs have been reviewed in the literature.10 CD30 (Ki-1 antigen) is a cytoplasmic and membrane-bound antigen identified on MCs at varying frequency in early and advanced forms of SM and has been used as an optional IHC and flow marker in SM studies.11,12 More recently, IHC and flow cytometric expression of programmed death-ligand 1 was identified in SM, providing an opportunity to explore immune checkpoint blockade in these neoplasms.13
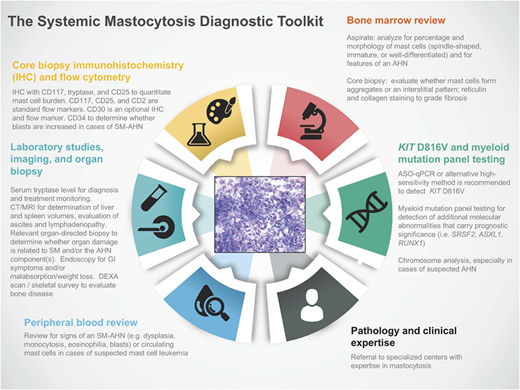
The SM diagnostic toolkit. Establishing a diagnosis of SM and staging the extent of disease require integrated clinical, histopathologic, laboratory, radiology, and molecular/cytogenetic testing. Referral to specialized centers with expertise in mastocytosis is recommended given the rarity of these diseases, particularly advSM variants. AHN, associated hematologic neoplasm; ASO-qPCR, allele-specific oligonucleotide quantitative polymerase chain reaction; CT, computed tomography; DEXA, dual-energy x-ray absorptiometry; GI, gastrointestinal; MRI, magnetic resonance imaging.
The SM diagnostic toolkit. Establishing a diagnosis of SM and staging the extent of disease require integrated clinical, histopathologic, laboratory, radiology, and molecular/cytogenetic testing. Referral to specialized centers with expertise in mastocytosis is recommended given the rarity of these diseases, particularly advSM variants. AHN, associated hematologic neoplasm; ASO-qPCR, allele-specific oligonucleotide quantitative polymerase chain reaction; CT, computed tomography; DEXA, dual-energy x-ray absorptiometry; GI, gastrointestinal; MRI, magnetic resonance imaging.
SM-AHN
Histopathologic evaluation of SM-AHN merits special consideration because of its associated diagnostic complexity and potential impact on therapeutic decision making. Lack of experience and/or the inappropriate use of antibodies may contribute to misdiagnosis or diagnostic delay of the SM and/or associated hematologic neoplasm (AHN) components in individual patients. Although a “catch-all” approach would be to check the serum tryptase level in all patients diagnosed with a myeloid neoplasm or have pathologists routinely use antibodies for tryptase and/or CD117, the added value and yield of this approach have not been well studied. At a minimum, the peripheral blood smear should be evaluated for signs of an AHN (ie, dysplasia, monocytosis, eosinophilia, and blasts); this may “prime” suspicion for BM involvement by an associated myeloid neoplasm (or rarely, a lymphoid neoplasm), such as myelodysplastic syndrome (MDS), MPN, MDS/MPN (most commonly chronic myelomonocytic leukemia [CMML]), chronic eosinophilic leukemia (CEL), not otherwise specified, or acute myeloid leukemia (AML). A large burden of MCs in the BM can mask a concomitant AHN, such as MDS, wherein dysplasia may only become apparent after cytoreduction of the SM component. The converse is also true; in some cases of systemic mastocytosis with acute myeloid leukemia (SM-AML), for example, previously unrecognized aggregates of spindled MC may only reveal themselves after a postinduction BM biopsy shows adequate hypoplasia and clearance of myeloblasts. In SM-AHN cases, it can be challenging to attribute organ damage to the SM or AHN component. Organ-directed biopsy is encouraged in such cases to guide treatment, because heterogeneity of organ involvement by the SM and AHN components is not uncommon in the same patient.
In the Mayo series, the median OS of SM-AHN patients was 2 years.4 Although the prognosis of SM-AHN frequently relates to the AHN component, marked heterogeneity in outcomes exists between AHN subtypes (ie, average survivals of 31, 15, and 13 months for SM-MPN, SM-CMML, and SM-MDS, respectively).7,14 Patients presenting with eosinophilia should be screened for the FIP1L1-PDGFRA fusion gene (fluorescence in situ hybridization for the CHIC2 gene deletion is commonly used), which defines a WHO disease entity distinct from SM.15,16 However, clinicopathologic findings in patients with the FIP1L1-PDGFRA fusion gene can mimic those with SM, including elevated serum tryptase levels, except that MCs typically show an interstitial pattern on the BM core biopsy.17 These patients, who are almost uniformly male, show exquisite sensitivity to imatinib.15-17
KIT D816V and myeloid mutation testing
The somatic D816V mutation in exon 17 of KIT is a major driver of SM pathogenesis.18 Multilineage involvement by KIT D816V is associated with disease progression and advanced disease.19 Allele-specific oligonucleotide quantitative polymerase chain reaction (ASO-qPCR; sensitivity ∼0.01%) or alternative high-sensitivity methods can detect KIT D816V in the peripheral blood or BM in >80% to 90% of patients with suspected SM.18,20 A higher KIT D816V VAF is seen in patients with advSM compared with ISM, which could be related to multilineage involvement and/or extensive disease burden.21 In ∼5% to 10% of patients, no KIT D816V mutation is detected. This may be due to the following reasons: (1) patients are, in fact, KIT D816V positive, but the (very) low MC burden leads to a false negative result, because the sensitivity of the assay is too low and/or the tissue sample is suboptimal (testing a BM sample is recommended if initial PCR fails to detect the KIT D816V mutation in the peripheral blood, especially in patients with ISM)21 ; (2) patients indeed only bear wild-type KIT; and (3) patients carry other rare KIT mutations in exon 17 that involve codon 816 (ie, D816F/H/I/Y), other codons (ie, D820G or N822K), or other regions of the KIT gene that are not detectable by the ASO-qPCR assay for KIT D816V.18 In patients with a negative KIT D816V screen by ASO-qPCR, screening of KIT for alternative codon 816 mutations requires amplification of exon 17 and sequencing of the resulting amplicons or preferably, peptide nucleic acid–mediated PCR.18 If no mutation is found at codon 816, sequencing of the whole KIT coding sequence by next generation sequencing (NGS) may be undertaken. However, the sensitivity of myeloid gene mutation panels for detection of KIT mutations is only 5% to 10% and may fail to detect low VAF.
We use myeloid mutation gene panel testing on the BM sample (unless patients have AHN and/or circulating MCs, where peripheral blood can be used) as an adjunctive diagnostic tool to assess for codriver molecular abnormalities that carry prognostic significance. The most commonly involved genes are TET2, SRSF2, ASXL1, RUNX1, JAK2, N/KRAS, CBL, and EZH2. One study identified additional molecular aberrations beyond KIT D816V in 24 of 27 (89%) patients with advSM (ASM, SM-AHN, and MCL) compared with 3 of 12 patients with ISM or SSM.22 TET2 mutations have a variable prevalence in SM,22-24 with hematopoietic colony assays showing that they often precede acquisition of the KIT D816V.25 KIT D816V and/or these additional molecular lesions may be coexpressed in the same MCs, and they may also be detected in other myeloid lineages, often in SM-AHN with a concurrent MDS or MDS/MPN.19,25,26 Mutations other than KIT D816V are rarely found in ISM, which is primarily driven by KIT D816V alone.22,25 The possibility of clonal hematopoiesis of indeterminate potential should be considered when interpreting myeloid gene panels, especially in patients who lack diagnostic findings of a concurrent WHO-defined hematologic neoplasm.
The prognostication toolkit
Inferior OS in SM has been associated with mutations in SRSF2, ASXL1, and/or RUNX1 (S/A/R panel),27 the occurrence of ASXL1 and/or CBL mutations,28 and the number of non-KIT 816V mutations.22,27,28 Even among the already poor-risk patients with MCL, S/A/R positivity remained the only independent poor prognostic variable predicting OS in a multivariate analysis.29 Among SM patients, abnormal karyotypes are typically found only in patients with SM-AHN. In 1 study, an abnormal cytogenetic profile was identified in 16 of 73 (22%) SM-AHN patients; among these 16 individuals, additional somatic mutations were identified in 12 (75%) patients.30 Seven of 10 (70%) patients with a poor-risk karyotype (ie, monosomy 7 or complex karyotype) and 1 of 6 (17%) patients with a good-risk karyotype progressed to secondary AML (n = 7) or MCL (n = 1) within a median of 40 months. The median OS of poor-risk karyotype patients was significantly shorter than that of good-risk/normal karyotype patients (4 vs 39 months; P < .0001), and it was independent of mutation status.
Clinical and genetic information has been combined to develop prognostic scoring systems that improve risk stratification in advSM. Spleen volume ≥450 mL on magnetic resonance imaging and elevated level of serum alkaline phosphatase were both associated with inferior survival in a multivariate analysis that included both ISM and advSM patients.31 Among advSM patients, only increased serum alkaline phosphatase level and mutation(s) in the S/A/R gene panel retained statistical significance in a multivariate analysis. The 3-year OS was 76% for patients with 0 to 1 (intermediate risk; n = 28) features and 38% for patients with 2 (high risk; n = 32) parameters.31 In the analysis of patients with advSM, age >60 years, hemoglobin <10 g/dL or transfusion dependence, platelet count <150 × 109/L, serum albumin <3.5 g/dL, and mutation in ASXL1 were each associated with inferior survival in a multivariate analysis.32 A “mutation-augmented prognostic scoring system” stratified these patients with advSM into high-, intermediate-, and low-risk groups with median survivals of 5, 21, and 86 months, respectively.32 Larger data sets of patients with advSM with annotated clinical, cytogenetic, and molecular information are being analyzed to validate and further refine risk stratification.
The treatment toolkit
In all subtypes of SM, avoidance of known triggers of MC activation is important. Patients should carry adrenaline autoinjectors at all times in case of anaphylaxis. Venom immunotherapy is used for long-term management of anaphylaxis in patients with evidence of hymenoptera-specific IgE or skin test positivity. Stepwise treatment approaches for MC mediator–related symptoms (ie, antihistamines, MC stabilizers, leukotriene receptor antagonists, corticosteroids, ketotifen, and the IgE-depleting antibody omalizumab) and bone disease (ie, bisphosphonates for osteoporosis and/or lytic lesions) have been extensively reviewed.33,34 For pregnancy or surgery planning, a proactive care plan generated by a multidisciplinary team of specialists, including anesthesia, allergy, high-risk obstetrics, and surgery, is recommended.35
For patients with advSM, cytoreductive therapy is used to reduce or reverse neoplastic MC–related organ damage. Figure 2 shows a treatment algorithm for advSM patients, including individuals with SM-AHN, in whom the primary decision making is whether treatment of the SM or AHN component takes immediate priority. For advSM patients, new consensus response criteria have been published by the International Working Group–Myeloproliferative Neoplasms Research and Treatment–European Competence Network on Mastocytosis (IWG-MRT-ECNM) (Figure 3).36 These criteria, which are anchored to reversion of organ damage and improvement of markers of MC burden (ie, serum tryptase levels and percentage BM MCs), were designed to harmonize response adjudication across clinical trials and have been adopted by regulatory health agencies.36
![Figure 2. Initial treatment considerations for advSM variants. Therapeutic options for patients with newly diagnosed ASM, SM-AHN, and MCL are shown. We always encourage participation in clinical trials. Midostaurin is Food and Drug Administration and European Medicines Agency approved for all 3 advSM subtypes, irrespective of KIT mutation status. For patients with SM-AHN, a determination must first be made regarding whether treatment is needed and which component requires more immediate therapy (in the text). Cladribine and [pegylated]–interferon-α with or without prednisone have been used on an off-label basis for all 3 advSM variants; cladribine is preferred in patients requiring rapid debulking of disease, whereas interferons are preferred in slow progressors. Imatinib is approved for the rare ASM patients who are KIT D816V mutation negative or whose KIT mutation status is unknown. For patients with eosinophilia, rule out FIP1L1-PDGFRA positivity, because such neoplasms are very sensitive to imatinib. Allogeneic hematopoietic stem cell transplantation (HSCT) may be considered as initial therapy for ASM or SM-AHN. We do not recommend transplantation as frontline treatment of MCL, but it may be considered for individuals achieving substantial clinical benefit with midostaurin, cladribine, or a clinical trial drug. We also do not recommend multiagent chemotherapy as initial treatment of MCL given the activity of these other agents. Corticosteroids may be used on a short-term basis for rapidly progressive SM-related organ damage.](https://ash.silverchair-cdn.com/ash/content_public/journal/hematology/2018/1/10.1182_asheducation-2018.1.127/2/m_hem01817f2.png?Expires=1767725099&Signature=Uc-8UWBcD6VPKu1r5B3TZ2hx-p~QhU-1NNXZSIk7GCj4wc9QaR4sdMaL2025Wp5JX7qZycz1~MGkpqRUHsF~Y4zjEYjLKzHyIp~0GTZTI6r3B03ReIP-UPsoAFiA8~gIdBEB-iN-w7odPcwF8uxudeOTjlycXEEDZOQkhGb6PpS~vUcbEbDdRHxKivCeqHQJfPb0N-7SCeHEHzhlrtBmlyiiVUpYp5ROv3QbfRRpm1LqGBlRYWp2YmxlqFnnLHCPCweqDzzOfTar3SMk~yLTPMjseE09d7zyQ8RmNypEDEUVaIsEbNQwL8bprq6XRGX1Uw40-jDu-iTo3bAyk~q4dQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Initial treatment considerations for advSM variants. Therapeutic options for patients with newly diagnosed ASM, SM-AHN, and MCL are shown. We always encourage participation in clinical trials. Midostaurin is Food and Drug Administration and European Medicines Agency approved for all 3 advSM subtypes, irrespective of KIT mutation status. For patients with SM-AHN, a determination must first be made regarding whether treatment is needed and which component requires more immediate therapy (in the text). Cladribine and [pegylated]–interferon-α with or without prednisone have been used on an off-label basis for all 3 advSM variants; cladribine is preferred in patients requiring rapid debulking of disease, whereas interferons are preferred in slow progressors. Imatinib is approved for the rare ASM patients who are KIT D816V mutation negative or whose KIT mutation status is unknown. For patients with eosinophilia, rule out FIP1L1-PDGFRA positivity, because such neoplasms are very sensitive to imatinib. Allogeneic hematopoietic stem cell transplantation (HSCT) may be considered as initial therapy for ASM or SM-AHN. We do not recommend transplantation as frontline treatment of MCL, but it may be considered for individuals achieving substantial clinical benefit with midostaurin, cladribine, or a clinical trial drug. We also do not recommend multiagent chemotherapy as initial treatment of MCL given the activity of these other agents. Corticosteroids may be used on a short-term basis for rapidly progressive SM-related organ damage.
Initial treatment considerations for advSM variants. Therapeutic options for patients with newly diagnosed ASM, SM-AHN, and MCL are shown. We always encourage participation in clinical trials. Midostaurin is Food and Drug Administration and European Medicines Agency approved for all 3 advSM subtypes, irrespective of KIT mutation status. For patients with SM-AHN, a determination must first be made regarding whether treatment is needed and which component requires more immediate therapy (in the text). Cladribine and [pegylated]–interferon-α with or without prednisone have been used on an off-label basis for all 3 advSM variants; cladribine is preferred in patients requiring rapid debulking of disease, whereas interferons are preferred in slow progressors. Imatinib is approved for the rare ASM patients who are KIT D816V mutation negative or whose KIT mutation status is unknown. For patients with eosinophilia, rule out FIP1L1-PDGFRA positivity, because such neoplasms are very sensitive to imatinib. Allogeneic hematopoietic stem cell transplantation (HSCT) may be considered as initial therapy for ASM or SM-AHN. We do not recommend transplantation as frontline treatment of MCL, but it may be considered for individuals achieving substantial clinical benefit with midostaurin, cladribine, or a clinical trial drug. We also do not recommend multiagent chemotherapy as initial treatment of MCL given the activity of these other agents. Corticosteroids may be used on a short-term basis for rapidly progressive SM-related organ damage.

IWG-MRT-ECNM response criteria for advSM. Normalization of 1 or more signs of hematologic and/or nonhematologic SM-related organ damage defines clinical improvement (CI). Refer to the published criteria (36 ) that define the type and severity of SM-related organ damage that is eligible for CI assessment as well as specific criteria for progressive disease (PD). The IWG-MRT-ECNM definitions of “eligible organ damage findings” are distinct from the WHO definitions of “C findings.” Attainment of partial remission (PR) and complete remission (CR) is based on increasing improvement of the serum tryptase level and BM MC burden. For CI, PR, and CR, response duration must be at least 12 weeks. *Changes in MC burden refer to the BM and/or extracutaneous organ(s). ANC, absolute neutrophil count; diff, differential; Hb, hemoglobin; PD, progressive disease; SD, stable disease.
IWG-MRT-ECNM response criteria for advSM. Normalization of 1 or more signs of hematologic and/or nonhematologic SM-related organ damage defines clinical improvement (CI). Refer to the published criteria (36 ) that define the type and severity of SM-related organ damage that is eligible for CI assessment as well as specific criteria for progressive disease (PD). The IWG-MRT-ECNM definitions of “eligible organ damage findings” are distinct from the WHO definitions of “C findings.” Attainment of partial remission (PR) and complete remission (CR) is based on increasing improvement of the serum tryptase level and BM MC burden. For CI, PR, and CR, response duration must be at least 12 weeks. *Changes in MC burden refer to the BM and/or extracutaneous organ(s). ANC, absolute neutrophil count; diff, differential; Hb, hemoglobin; PD, progressive disease; SD, stable disease.
Interferon-α and cladribine
Interferon-α (IFN-α) and cladribine have historically been used on an off-label basis as first-line agents for advSM, with the latter favored in patients with kinetically active disease who require rapid debulking of MCs. [PEG]–IFN-α with or without oral steroids has been evaluated in small retrospective series that have often included a heterogeneous admixture of indolent and advSM patients with limited follow-up, confounding interpretation of response.37,38 IFN-α treatment has shown variable success in diminishing skin findings and symptoms, improving osteoporosis, and ameliorating cytopenias. The time to response may approach a year, and inconsistent reduction in MC burden and relatively rapid relapse after discontinuation suggest cytostatic effects on MCs. Toxicities associated with IFN-α use may include flu-like symptoms, myelosuppression, transaminitis, thyroid dysfunction, autoimmune phenomena, and depression, which can impact tolerance and long-term use.37,38
Cladribine (2-chlorodeoxyadenosine) has been studied in indolent and advanced subtypes of SM. The long-term retrospective French experience in both indolent (36 patients) and advSM (32 patients) patients was recently reported, and this is the largest series of cladribine to date.39 Cladribine was administered at a dose of 0.14 mg/kg as a 2-hour intravenous infusion or subcutaneously for 5 days every 4 to 12 weeks. A median of 3.7 courses was administered (range, 1-9). The overall response rate (ORR) was 72%; 92% of ISM patients responded compared with 50% of patients with advanced disease. Among patients with advSM, the complete response (CR), major response (MR), and partial response (PR) rates were 0%, 37.5%, and 12.5%, respectively. No response was observed in the 1 patient with MCL. Significant decreases in serum tryptase levels were only observed in ISM patients, and changes in BM MC burden were evaluated only in 9 patients, making it difficult to draw conclusions regarding this end point. The median duration of response (DOR) was 2.47 (0.5-8.6) years for advSM. Lymphopenia (82%), neutropenia (47%), and opportunistic infections (13%) were the most common grade 3/4 adverse events (AEs).
Tyrosine kinase inhibitors
Imatinib is a multikinase inhibitor that is active against wild-type, but not D816V-mutated, KIT.40 Imatinib was approved by the US Food and Drug Administration in 2006 for adult patients with ASM without the D816V KIT mutation or with unknown KIT mutational status. With the advent of D816V ASO-qPCR, the pool of patients without KIT D816V mutation has decreased, and the utility of imatinib is limited to those with wild-type KIT or rare nonexon 17 mutations (eg, F522C; germ line K509I; deletion of codon 419 in exon 8; p.A502_Y503dup in exon 9),41,42 some of which show well-differentiated MC morphology (ie, round MCs; low/absent CD25) in contrast to the spindled neoplastic MC morphology with CD25 expression.43
Although dasatinib and nilotinib exhibit in vitro activity against MCs carrying the KIT D816V mutation, these agents have shown low ORRs in open label phase 2 trials44,45 and are no longer being pursued in advSM.
Midostaurin is a multikinase inhibitor that targets wild-type and D816V-mutated KIT in addition to other protein kinases, such as FLT3, PDGFR-α/β, and VEGFR2. In Ba/F3 cells transformed by KIT D816V, the 50% inhibitory concentration (IC50) of midostaurin was 30 to 40 nM compared with >1 μM with imatinib.46 In a phase 2 investigator-initiated trial, 100 mg midostaurin twice daily was administered to 26 patients with advSM (ASM [n = 3], SM-AHN [n = 17], and MCL [n = 6]) with 1 or more C findings.47 ORR and MR were 69% and 50%, respectively, with 68% of evaluable patients having a decreased MC burden in the BM by ≥50%. Responses were durable, with 2 patients achieving complete remission on long-term follow-up, including a patient with MCL and unmasked MDS carrying KIT D816V and FLT3 mutations. The median OS and progression-free survival (PFS) were 40 and 41 months, respectively.
These encouraging results led to a global, single-arm, open label registration trial of midostaurin that included 116 patients, of whom 89 advSM patients with ≥1 C findings were evaluable for response (ASM [n = 16], SM-AHN [n = 57], and MCL [n = 16]).48 The trial showed ORR and MR of 60% and 45%, respectively, per modified Valent and Cheson criteria. Responses were observed regardless of KIT D816V status, prior therapy, or the presence of an AHN. The median best reduction in serum tryptase level was –58%. In addition, the median change in BM MC burden was –59%, and 57% of patients had a ≥50% reduction in the BM MC burden. Spleen volume decreased in 77% of evaluable patients. After a median follow-up of 26 months, the median DOR, the median OS, and the median PFS were 24.1, 28.7, and 14.1 months, respectively. Median OS in responders was 44.4 months. Of the 16 patients with MCL, 8 responded, including 7 with MR (44%), and the median DOR was not reached. In a post hoc multivariate analysis, outcomes that were associated with longer OS included achievement of a response and a ≥50% decrease in BM MC burden. Eleven percent of patients (almost all with SM-AHN at baseline) progressed to secondary AML, which is the same rate observed in a Mayo Clinic natural history study.4,48
Midostaurin was generally well tolerated. Most AEs were gastrointestinal in nature: mainly nausea and to a lesser extent, vomiting and diarrhea (6%-8% grade 3/4). New or worsening grade 3/4 neutropenia, anemia, and thrombocytopenia occurred in 24%, 41%, and 29%, respectively, mostly in patients with preexisting cytopenias. Symptoms and quality of life were significantly improved with midostaurin treatment, except for nausea and vomiting, consistent with the drug’s tolerability profile. To mitigate nausea, prophylactic antiemetics are recommended before each dose of midostaurin as well as coadministration with food. The improvement in disease-related symptoms may relate to midostaurin’s combined inhibitory effects on MC proliferation and mediator release.49 Based on this pivotal trial, midostaurin was approved by the Food and Drug Administration and the European Medicines Agency in 2017 for the treatment of patients with advSM.
Biologic correlates of response and mechanisms of resistance to midostaurin in patients with advSM have not been fully elucidated. Secondary mutations in KIT have not been reported to date. The impact of additional molecular aberrations and KIT D816V VAF on the outcome of 38 patients with advSM treated with midostaurin was recently evaluated.50 Response rate and OS were significantly superior in (S/A/R) gene panel–negative compared with (S/A/R) gene panel–positive patients tested by NGS. In addition, patients with ≥25% reduction in KIT D816V VAF (KIT responders) by 6 months of treatment had a significantly higher response rate to midostaurin, longer duration of therapy, and improved OS compared with KIT nonresponders.50 Serial NGS panels were undertaken in 16 patients to investigate clonal dynamics on midostaurin. Among 7 patients with disease progression, there was an increasing VAF or emergence of new mutation(s) in K/NRAS, RUNX1, IDH2, or NPM1. Among the 8 patients with durable responses to midostaurin, there were 6 patients who had disappearance of detectable KIT D816V, CBL, and/or TET2 mutations. The remaining 2 patients died while on treatment, with increasing VAFs noted in KRAS and RUNX1; the latter was associated with transformation from SM-MDS/MPN to SM-AML. Two patients exhibited new JAK2 V617F mutations, including 1 individual who transformed from ASM to ASM-PV. These clonal changes occurred irrespective of the patients’ KIT D816V VAF.50
Avapritinib (BLU-285; Blueprint Medicines) is an oral type 1 multikinase inhibitor with highly selective and potent activity against KIT and PDGFRA A-loop mutants, including D816V (IC50 = 0.27 nM).51 Based on its significant activity in several preclinical models, including HMC1.2 cell lines (V560G and D816V mutated) and mice xenografted with P815 mastocytoma cells,51 a multicenter phase 1 trial with dose escalation and expansion phases was initiated in advSM patients (ClinicalTrials.gov identifier NCT02561988).
The results of the dose escalation and expansion phases have been reported, with the expansion phase currently ongoing.52,53 Responses in organ damage, MC burden, and spleen volume were observed regardless of advSM subvariant, prior therapy, or mutational profile (eg, S/A/R status). Among 36 patients with BM MCs ≥5% and after baseline BM assessment and tryptase measurement, 29 (81%) experienced ≥50% reduction in both measures. In addition, 88% (37 of 42) of patients exhibited ≥50% reduction of KIT D816V VAF early in the course of treatment. Among the 52 enrolled patients in the dose escalation and expansion cohorts, 23 patients were evaluable for response by IWG-MRT-ECNM criteria.53 At the time of data cutoff, the ORR was 83%, consisting of 17% CR plus CR with partial recovery of peripheral blood counts, 53% PR, and 13% with clinical improvement. Responses were also observed in patients who had experienced progression or intolerance on prior midostaurin therapy. Periorbital edema, fatigue, nausea, diarrhea, peripheral edema, and cognitive effects were the most commonly reported AEs, primarily grades 1 and 2 in nature. Myelosuppression, including thrombocytopenia and anemia, as well as some cases of nausea, vomiting, fatigue, ascites, and periorbital edema were the most common grades 3 and 4 AEs. Dose reductions and interruptions due to AEs were reported in 56% of patients each, most of which occurred at doses of >200 mg daily. After median follow-up of 14 and 5 months in the dose escalation and expansion groups, respectively, 42 of 52 (80%) enrolled patients remained on treatment.52,53 The phase 1 dose expansion cohort continues enrollment at starting doses of 200 to 300 mg daily, and the phase 2 study has initiated accrual.
DCC-2618 (Deciphera Pharmaceuticals) is a type 2 switch control kinase inhibitor with activity against multiple KIT and PDGFR-α mutants, with antineoplastic activity in preclinical models using KIT WT- (IC50 = 11-61 nM) and D816V-transfected (IC50 = 133-256 nM) MC lines.54 Enrollment of patients with advSM as part of an expanded phase 2 trial is in progress with a recommended phase 2 dose of 150 mg daily (ClinicalTrials.gov identifier NCT02571036).
Allogeneic hematopoietic stem cell transplantation
The outcome of allogeneic hematopoietic stem cell transplantation (HSCT) in advSM has not been studied prospectively. A multicenter retrospective study showed that median OS at 3 years was 57% for all patients, 74% for patients with SM-AHN, and 43% and 17% for ASM and MCL patients, respectively.55
The strongest risk factor for worse OS was a diagnosis of MCL, and lower OS was observed in patients undergoing reduced intensity vs myeloablative conditioning. Treatment-related mortality was 20% at 1 year.55 Currently, we refer patients for transplant whenever it is indicated for the AHN component (eg, AML, high-risk MDS, MDS/MPN). For patients with an available donor who are exhibiting high-quality responses to midostaurin or a selective KIT D816V inhibitor, no data are available to guide whether such individuals should be continued on therapy or be referred for transplant. Consensus opinion on HSCT in advSM was recently published.56 These guidelines will be further refined by integrating long-term follow-up data from the KIT inhibitor trials and prognostic scoring systems that incorporate molecular profiling.
A clinical trials roadmap for advSM
With the approval of midostaurin and encouraging early-phase data with avapritinib, KIT inhibition is now a validated therapeutic approach in advSM. Although more mature data need to emerge from the second generation of selective KIT D816V inhibitors, a clinical trials roadmap can be envisioned for advSM (Figure 4). It is unclear whether KIT inhibitors are exerting any meaningful impact on AHNs, despite the frequent co-occurrence of KIT D816V in cells of the AHN or the clinical observation of improvement or resolution of peripheral blood monocytosis or eosinophilia (ie, in cases of SM-CMML or SM with CEL, respectively). This likely relates to the genetic complexity of the AHNs beyond KIT D816V, and it should encourage the development of basket-type trials evaluating combinations of KIT inhibitors with agents approved for specific AHNs or the selection of agents based on actionable targets identified on mutation panels (Figure 4). Such studies can inform protocols of specific doublets for particular SM-AHNs.

A clinical trials roadmap for advSM. Over the next few years, several avenues of clinical trial investigation are envisioned based on unmet needs of this patient population and the availability of new therapeutics. HSC, hematopoietic stem cell.
A clinical trials roadmap for advSM. Over the next few years, several avenues of clinical trial investigation are envisioned based on unmet needs of this patient population and the availability of new therapeutics. HSC, hematopoietic stem cell.
Based on preclinical data showing synergistic activity, a potential role exists for a syncopated regimen of cladribine plus KIT inhibition in advSM patients with refractory/relapsed disease to KIT inhibitor monotherapy. Similar to the questions raised with JAK inhibitors in myelofibrosis,57 additional studies are needed to understand how KIT inhibition may be used as a bridging strategy in the pretransplant period and after transplant to reduce the risk of relapse.
Back to the patient scenario
The patient agrees to a liver biopsy that shows a marked, multifocal, dense hepatic inflammation with clusters of CD117- and CD25-positive MCs in a portal, periportal, and lobular distribution. A rereview of the patient’s initial diagnostic marrow from 18 months earlier showed PMF but overlooked a similar infiltrate of neoplastic MCs comprising 10% of the marrow cellularity. The patient was given a revised diagnosis of SM-PMF. The KIT D816V VAF of 38% is relatively high for the low BM MC of 10% and suggests the presence of an SM-AHN with multilineage involvement of the mutation.
The patient’s increased alkaline phosphatase and paracentesis-dependent ascites were assessed as organ damage (C findings) related to neoplastic MC infiltration of the liver. The 10% marrow MC infiltrate is an unlikely explanation for the cytopenias; however, ineffective hematopoiesis due to PMF (and fibrosis) and hypersplenism (from the SM and/or PMF components) may also be contributory. We prioritized treatment of the SM component given the nascent liver-related complications. We discussed enrollment in a clinical trial with avapritinib or treatment with midostaurin; he chose the latter because of the challenge of long-distance travel for study visits. Depending on his response, we will also revisit the role of allogeneic HSCT.
Conclusion and future directions
KIT inhibition has assumed a central role in the treatment of advSM, and it has reenergized interest in developing the requisite tools to optimize patient diagnosis, risk stratification, and response assessment. The application of NGS myeloid mutation panels and KIT D816V VAF monitoring is helping to reshape our understanding of the clonal landscape of these diseases as well as the dynamics of treatment response and progression. To inform the clinical trials roadmap, new drug development will be bundled with correlative biomarkers, such as circulating tumor DNA, multiplex cytokine profiling, and RNA sequencing of bulk or single cells comprising the SM and AHN components, to better characterize the disease and treatment effect. These precision medicine tools may also permit more individualized treatment. Progress in these rare diseases is critically dependent on collaborations between MC-focused academic organizations (eg, the European Competence Network on Mastocytosis and the newly formed American Initiative in Mast Cell Diseases), patient advocacy groups (eg, The Mastocytosis Society), and biopharma. Dissemination of recently developed National Comprehensive Cancer Network guidelines for SM should help in the adoption of consensus algorithms for the diagnosis and treatment of these challenging disorders.58
Acknowledgments
The authors thank the Charles and Ann Johnson Foundation for their support and the members of the European Competence Network on Mastocytosis for their collaborative efforts in mastocytosis research.
Correspondence
Jason Gotlib, Medicine (Hematology), Stanford Cancer Institute, 875 Blake Wilbur Dr, Stanford, CA 94305-6555; e-mail jason.gotlib@stanford.edu.
