
Abstract
Mutational profiling has fundamentally changed our approach to patients with acute myeloid leukemia (AML). Patients with AML are routinely profiled for the presence of mutations in FLT3, NPM1, CEBPA, and, more recently, TP53. In this chapter, we review the role of mutational profiling to help define disease biology in AML, particularly among patients with putatively intermediate-risk disease. We describe the body of evidence supporting the utility of mutational profiling when performed at the time of diagnosis (to identify prognostic and targetable mutations), at the time of complete remission (to assess minimal residual disease as a marker for relapse), and at the time of relapse (to identify therapeutic targets and eligibility for clinical trials). We further identify particular mutations that have been shown to affect prognosis across the established European LeukemiaNet risk categories and discuss which mutational events might be used to alter the approach to patient care at various time points during the disease course. We also review the evidence in support of molecular profiling for assessment of minimal/measurable residual disease and describe the current landscape of studies designed to validate this approach.
Learning Objectives
Learn how mutations can add additional prognostic information to classification within the European LeukemiaNet risk assessment for newly diagnosed AML
Describe the potential role of molecular profiling at diagnosis, in remission, and at relapse and how profiling at each of these times helps to modify the approach to treatment of patients with AML
Introduction
The most common type of acute leukemia in adults is acute myeloid leukemia (AML), which presents at a median age of 68 years.1 In 2018, ∼20 000 people will be diagnosed with AML, and ∼11 000 people will die as a result of the disease, accounting for ∼1% of US cancer diagnoses and 2% of cancer deaths.1 Despite substantial overlap in clinical presentation across the different World Health Organization subtypes of AML, optimal management of adults with AML is complicated by the relatively recent recognition of molecular disease subsets (distinct from the well-recognized contribution of cytogenetic risk) with different responses to standard therapeutics. These results are further confounded by the seemingly different contribution of select molecular events in older vs younger patients and the relatively limited data regarding response prediction for patients receiving nonintensive clinical therapy.2-6 In recognition of the role of molecular events in AML, the European LeukemiaNet (ELN) revised the prognostic tool for leukemia in 2017 to add mutations in RUNX1, ASXL1, and BCR-ABL1 to the previously identified molecular risk categories defined by mutations in NPM1, CEBPA, FLT3-ITD, and TP53.6 This tool stratifies patients using cytogenetic and molecular features into 3 prognostic groups (good, intermediate, and poor risk) based upon predicted response to standard therapy and survival (Table 1).6 A majority of commercial mutational profiles require a 1- to 2-week turnaround; therefore, panel results are unlikely to be available during the selection of upfront therapy. For select genes, including FLT3 and perhaps IDH1/2 mutations, more expeditious test methodologies may be warranted to allow for upfront incorporation of targeted therapeutics. Although incorporation of these new mutational events has improved upon prior prognostic tools,7,8 many more genes are likely to contribute to leukemia pathogenesis as well as to potentially inform optimal therapeutics (including the use of established and new agents; Table 1).2,9 Complicating things further, recent work has uncovered that the AML epigenome adds an entirely new layer of complexity, with its own heterogeneous subsets that function independently of AML genetic diversity, a conundrum beyond the scope of this review.10
Incorporating cooccurring mutational data into 2017 ELN risk strata
| ELN 2017 category | Genetic abnormality | Concurrent mutation frequency | Protective | Deleterious |
|---|---|---|---|---|
| Favorable | t(8;21)(q21;q22.1); RUNX1-RUNX1T1 (7%) | KIT 25%, NRAS ∼20%, cohesin ∼20%, | KIT, FLT3 increase relapse risk, | |
| ZBTB7A ∼20%, ASXL1* ∼10% | NRAS, KRAS decrease relapse risk26 ; cohesin/chromatin modifier genes predict adverse outcomes26 ; cooccurrence of >1 TK/RAS gene increases relapse risk28 | |||
| inv(16)(p13.1;q22)/t(16;16)(p13.1;q22) CBFB-MYH11 (5%) | NRAS 40%, KIT 35%, FLT3 TKD 20%, KRAS 15% | NRAS, KRAS decrease relapse risk26 | KIT/FLT3 increase relapse risk | |
| Cooccurrence of >1 TK/RAS gene increases relapse risk28 | ||||
| NPM1mutFLT3-ITDlow-wt | DNMT3A 50%, FLT3-ITD 40%, cohesin ∼20%, NRAS 20%, IDH1 15%, IDH2 R140 15%, TET2 15% | Cooccurrence of chromatin modifier mutations associated with favorable risk2 | Cooccurrence of NPM1, DNMT3A, FLT3 ITD mutations predicts poor prognosis12 | |
| Biallelic CEBPA(4%) | GATA2∼30%, NRAS∼30%, WT1 ∼20%, CSF3R∼20% | Germ line biallelic CEBPA-mutant patients at significant risk of postremission relapse and donor-derived leukemia33 | ||
| Intermediate | NPM1mutFLT3-ITDhigh | Mutations in chromatin modifiers (STAG2, BCOR, MLLPTD, EZH2, and PHF6) and RNA splicing factors (SRSF2, SF3B1, U2AF1, and ZRSR2) associated with MDS-like disease biology and poor outcome | ||
| NPM1wtFLT3-ITDlow-wt | ||||
| t(9;11)(p21.3;q23.3); MLLT3-KMT2A | ||||
| NOS favorable/adverse | ||||
| Adverse | t(6;9)(p23;q34.1); DEK-NUP214 (1%) | FLT3-ITD ∼70%, KRAS ∼20% | ||
| t(9;22)(q34.1;q11.2); BCR-ABL1 (1%) | ||||
| inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2); GATA2, MECOM(EVI1) (1%) | NRAS ∼30%, KRAS ∼15%, PTPN11 ∼20%, SF3B1 ∼20%, GATA2 15%, ETV6 5%, PHF6 ∼15%, BCOR ∼10%, RUNX1 ∼10%, ASXL1 ∼10%, NF1∼10% | |||
| t(v;11q23.3); KMT2A rearranged (4%) | KRAS ∼20%, NRAS ∼20% | |||
| −5 or del(5q); −7; −17/abn(17p) | ||||
| TP53mut or complex/monosomal (≥2 monosomies) karyotype 10% | BRCA1, BRCA2, CHEK2, PALB2 in up to 21% of patients with secondary AML40,41 | |||
| NMP1wtFLT3-ITDhigh | Adverse prognosis mitigated with upfront allo-SCT42,43 | |||
| RUNX1mut* | ||||
| ASXL1mut* |
| ELN 2017 category | Genetic abnormality | Concurrent mutation frequency | Protective | Deleterious |
|---|---|---|---|---|
| Favorable | t(8;21)(q21;q22.1); RUNX1-RUNX1T1 (7%) | KIT 25%, NRAS ∼20%, cohesin ∼20%, | KIT, FLT3 increase relapse risk, | |
| ZBTB7A ∼20%, ASXL1* ∼10% | NRAS, KRAS decrease relapse risk26 ; cohesin/chromatin modifier genes predict adverse outcomes26 ; cooccurrence of >1 TK/RAS gene increases relapse risk28 | |||
| inv(16)(p13.1;q22)/t(16;16)(p13.1;q22) CBFB-MYH11 (5%) | NRAS 40%, KIT 35%, FLT3 TKD 20%, KRAS 15% | NRAS, KRAS decrease relapse risk26 | KIT/FLT3 increase relapse risk | |
| Cooccurrence of >1 TK/RAS gene increases relapse risk28 | ||||
| NPM1mutFLT3-ITDlow-wt | DNMT3A 50%, FLT3-ITD 40%, cohesin ∼20%, NRAS 20%, IDH1 15%, IDH2 R140 15%, TET2 15% | Cooccurrence of chromatin modifier mutations associated with favorable risk2 | Cooccurrence of NPM1, DNMT3A, FLT3 ITD mutations predicts poor prognosis12 | |
| Biallelic CEBPA(4%) | GATA2∼30%, NRAS∼30%, WT1 ∼20%, CSF3R∼20% | Germ line biallelic CEBPA-mutant patients at significant risk of postremission relapse and donor-derived leukemia33 | ||
| Intermediate | NPM1mutFLT3-ITDhigh | Mutations in chromatin modifiers (STAG2, BCOR, MLLPTD, EZH2, and PHF6) and RNA splicing factors (SRSF2, SF3B1, U2AF1, and ZRSR2) associated with MDS-like disease biology and poor outcome | ||
| NPM1wtFLT3-ITDlow-wt | ||||
| t(9;11)(p21.3;q23.3); MLLT3-KMT2A | ||||
| NOS favorable/adverse | ||||
| Adverse | t(6;9)(p23;q34.1); DEK-NUP214 (1%) | FLT3-ITD ∼70%, KRAS ∼20% | ||
| t(9;22)(q34.1;q11.2); BCR-ABL1 (1%) | ||||
| inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2); GATA2, MECOM(EVI1) (1%) | NRAS ∼30%, KRAS ∼15%, PTPN11 ∼20%, SF3B1 ∼20%, GATA2 15%, ETV6 5%, PHF6 ∼15%, BCOR ∼10%, RUNX1 ∼10%, ASXL1 ∼10%, NF1∼10% | |||
| t(v;11q23.3); KMT2A rearranged (4%) | KRAS ∼20%, NRAS ∼20% | |||
| −5 or del(5q); −7; −17/abn(17p) | ||||
| TP53mut or complex/monosomal (≥2 monosomies) karyotype 10% | BRCA1, BRCA2, CHEK2, PALB2 in up to 21% of patients with secondary AML40,41 | |||
| NMP1wtFLT3-ITDhigh | Adverse prognosis mitigated with upfront allo-SCT42,43 | |||
| RUNX1mut* | ||||
| ASXL1mut* |
Columns 1 and 2 display the revised 2017 World Health Organization guidelines for AML risk stratification, with percentage of total AML cases in each subset in parentheses. Columns 3, 4, and 5 display mutations observed within each subset and their implications.
allo-SCT, allogeneic stem-cell transplantation; MDS, myelodysplastic syndromes; NOS, not otherwise specified; TK, tyrosine kinase; WT, wild type.
Mutations in RUNX1/ASXL1 do not imply adverse outcomes when they cooccur within an otherwise favorable-risk subtype.
Although the ELN 2017 prognostic model considers just 1 mutation in the majority of patients and 2 in a minority of others, patients with AML have an average of 3 acquired mutations (range, 0-9) at the time of diagnosis.2,11,12 Older patients have on average 1 more mutation than younger patients, with increasing numbers of mutational events associated with worsening prognosis.2,12 Comparison of various mutational tools for prediction of outcome has revealed that most can add significantly to the current prognostic models, but outside of a prospective clinical trial, it is difficult to know which tool to use and how to incorporate them into standard practice.8
Sequencing of older and younger adults without hematological malignancies and the extensive evaluation of mutational burden using variant allele frequencies (VAFs) in patients with myeloid malignancies have exposed the ontogenic hierarchy of acquired mutations and the oligoclonality in AML as substantial modifiers of disease relapse and resistance (Figure 1). These studies have helped to explain why detection of particular mutational events, such as those associated with CHIP at the time of CR, are not associated with outcome, whereas others are strongly predictive of relapse.13-17 On the basis of these insights, it is clear that serial evaluation of the molecular profile in patients with AML, most importantly at the times of diagnosis, remission, and relapse, can substantially improve our understanding of the disease process and potentially enhance our approach to patient management.
![Figure 1. The role of molecular profiling in AML diagnosis, therapeutic selection, and disease monitoring. In this schematic, AML development is depicted as the terminal stage in a phenotypic continuum from prior clonal hematopoiesis and myelodysplasia. A sample of acquired mutations (not all encompassing) driving disease progression is listed with respect to time (x-axis), in addition to a model for longitudinal molecular assessment. Relative clonal size is depicted on the y-axis. In this sample case, mutational profiling at diagnosis aids in selection of a therapy that leads to a significant reduction of leukemic burden and establishment of a first complete remission (CR). From here, 3 possible scenarios are depicted: sustained remission with expansion of preexisting nonmalignant clones (clonal hematopoiesis of indeterminate potential [CHIP],13-17 disease relapse with development and outgrowth of a leukemic subclone (blue),51 and control of persistent disease with targeted agents leading to cellular differentiation without killing (eg, IDH1/2 inhibitors, hypomethylating agents).4 IDH, isocitrate dehydrogenase; MRD, minimal residual disease.](https://ash.silverchair-cdn.com/ash/content_public/journal/hematology/2018/1/10.1182_asheducation-2018.1.35/2/m_hem01805f1.png?Expires=1767702851&Signature=ikipnPXFMiGkwQ3swgR8ZoFVvjSS-qT9vPX8t~gRcfoH3dkYEy~oXgzT-C5SvPm7A-osDG0MfdrZDrooosTX7ONHutO0OrJSF7YmkI-BzefFL6YQ5lxU~fB7rXpEIpoKsdTKNwLX6Z5616Ye9ZcxiTlI~RehtzH6yj2IQUa63y~esXY9pUfGuexYtQCsquCBKXI0C~lKmQL0bFiKKhn5MpC6ucyWNYqXX9v0OAZUntcmQsHYk25QIMgoFfstLReB9U4xlopFEHar2wgnj5qAoLLbboAZY49xAJS48ZSxXocGAz5NDPMRlYxQ2LkMXah9zTQZZmGPmi5Sh5KlrpuyEg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The role of molecular profiling in AML diagnosis, therapeutic selection, and disease monitoring. In this schematic, AML development is depicted as the terminal stage in a phenotypic continuum from prior clonal hematopoiesis and myelodysplasia. A sample of acquired mutations (not all encompassing) driving disease progression is listed with respect to time (x-axis), in addition to a model for longitudinal molecular assessment. Relative clonal size is depicted on the y-axis. In this sample case, mutational profiling at diagnosis aids in selection of a therapy that leads to a significant reduction of leukemic burden and establishment of a first complete remission (CR). From here, 3 possible scenarios are depicted: sustained remission with expansion of preexisting nonmalignant clones (clonal hematopoiesis of indeterminate potential [CHIP],13-17 disease relapse with development and outgrowth of a leukemic subclone (blue),51 and control of persistent disease with targeted agents leading to cellular differentiation without killing (eg, IDH1/2 inhibitors, hypomethylating agents).4 IDH, isocitrate dehydrogenase; MRD, minimal residual disease.
The role of molecular profiling in AML diagnosis, therapeutic selection, and disease monitoring. In this schematic, AML development is depicted as the terminal stage in a phenotypic continuum from prior clonal hematopoiesis and myelodysplasia. A sample of acquired mutations (not all encompassing) driving disease progression is listed with respect to time (x-axis), in addition to a model for longitudinal molecular assessment. Relative clonal size is depicted on the y-axis. In this sample case, mutational profiling at diagnosis aids in selection of a therapy that leads to a significant reduction of leukemic burden and establishment of a first complete remission (CR). From here, 3 possible scenarios are depicted: sustained remission with expansion of preexisting nonmalignant clones (clonal hematopoiesis of indeterminate potential [CHIP],13-17 disease relapse with development and outgrowth of a leukemic subclone (blue),51 and control of persistent disease with targeted agents leading to cellular differentiation without killing (eg, IDH1/2 inhibitors, hypomethylating agents).4 IDH, isocitrate dehydrogenase; MRD, minimal residual disease.
In this review, we discuss the relatively large body of data in support of molecular profiling for prognostication and therapeutic response assessment in younger and older adults with AML across the spectrum of ELN classifications.6 We also review the case for evaluation of MRD using molecular techniques in patients at the time of documented remission and touch on the use of molecular profiling to identify potentially targetable events in patients at the time of relapse.4,15,18-20
Mutational profiling in adults with AML
A majority of patients with AML will undergo upfront risk stratification with conventional karyotyping, with ∼50% of patients having a normal karyotype. Samples of bone marrow or peripheral blood containing blasts are appropriate for mutational profiling.21 Mutations in selected genes have been most important for those patients with a normal karyotype.2,11,22 Indeed, a majority of leukemia clinicians will now routinely test for mutations in NPM1, CEBPA, and FLT3. With the advent of the ELN 2017 guidelines, testing for RUNX1 (now included as a provisional entity), ASXL1, and TP53 (associated with inferior therapeutic response and survival12,23,24 ) should certainly be added to this list, but there are other mutational events (particularly those for which targeted therapy is now approved, such as IDH1/2) that are likely to be meaningful for the practicing leukemia physician (Table 1; Figure 1). In the next sections, we review the role of molecular profiling within each patient subgroup as defined by ELN 2017.
Age
Although cytogenetic and molecular profiling substantially contribute to survival prediction, age at diagnosis remains among the most powerful predictors of outcome. In this context, age may be acting as a surrogate for both underlying disease biology (chemotherapy resistance or secondary disease) and performance status.25 Molecular profiling may help us to untangle the former from the latter and in so doing allow the clinician to treat the patient rather than the numerical age. Figure 2 depicts the relative impact of molecular, cytogenetic, and clinical features as defined across a cohort of >1500 patients with AML.12 In this figure, the size of each bubble is indicative of the frequency of each occurrence within the cohort, and the location on the x-axis describes the effect size (protective or harmful to outcome). In this cohort of relatively younger patients (age range, 18-84 years; 89% age <60 years), age retained substantial adverse prognostic significance, whereas allografting was strongly protective. This figure highlights the relative impact of molecular and clinical features and helps put molecular profiling into context with familiar clinical variables. It is important to recognize that a majority of published data on the use of molecular profiling are biased toward younger patients. Despite this, several groups have confirmed that early identification of the presence of NPM1 mutations, the identification of potentially targetable mutations in FLT3 and other tyrosine kinases, and the recognition of poor-risk mutational events can help to better define prognosis in older patients.2,7,12,26 This is particularly important for older patients who are likely to derive substantial survival benefit from high-dose induction therapy and in whom performance status, baseline platelet count, albumin, and other more concrete clinical features may be superior to age in determining tolerance of therapy with curative intent.2,25 The identification of molecular events that define disease biology will help the clinician determine an optimal approach to management, intensive or not, in patients whose prior therapeutic options might have been limited by age alone.2,26
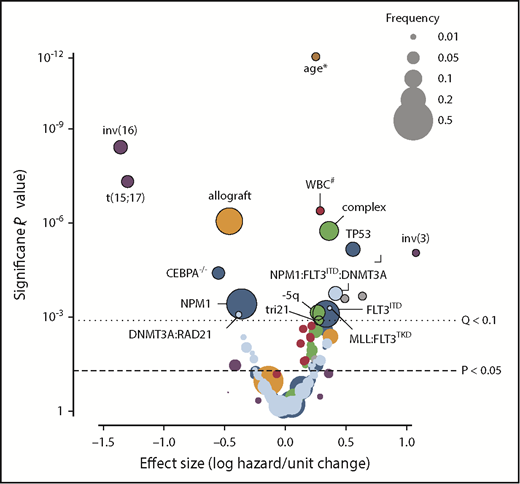
Outcomes in AML modified by clinical, cytogenetic, and molecular features as well as therapy. This volcano plot shows the relative contribution to prognosis (expressed as the logarithmic hazard on the x-axis; positive values indicate a worsening effect) vs P values (expressed on an inverted logarithmic scale on the y-axis) for each of 228 variables included in their random-effects model. Circles above the dotted line represent 18 variables with a q value of <0.1; the size of each circle corresponds to the frequency of the variable, as indicated in the box. The incremental contribution of age is shown for every 10 years of age, and the incremental contribution of the white blood cell count (WBC) is shown for each increase of 1 × 109 cells/L. Colors correspond to clinical variables (red), gene-gene interactions (light blue), copy number variations (green), treatment effect (orange), demographics (pink), and fusion genes (purple). Data reprinted with permission.12
Outcomes in AML modified by clinical, cytogenetic, and molecular features as well as therapy. This volcano plot shows the relative contribution to prognosis (expressed as the logarithmic hazard on the x-axis; positive values indicate a worsening effect) vs P values (expressed on an inverted logarithmic scale on the y-axis) for each of 228 variables included in their random-effects model. Circles above the dotted line represent 18 variables with a q value of <0.1; the size of each circle corresponds to the frequency of the variable, as indicated in the box. The incremental contribution of age is shown for every 10 years of age, and the incremental contribution of the white blood cell count (WBC) is shown for each increase of 1 × 109 cells/L. Colors correspond to clinical variables (red), gene-gene interactions (light blue), copy number variations (green), treatment effect (orange), demographics (pink), and fusion genes (purple). Data reprinted with permission.12
Germ line predispositions
The ELN 2017 guidelines now include a category of myeloid disorders with germ line predisposition. They identify germ line–predisposing events associated with mutations in CEBPA, DDX41, RUNX1, ANKRD26, ETV6, GATA2, TP53 (Li Fraumeni syndrome), the dyskeratosis congenita genes (TERC/TERT), and Fanconi anemia genes.6 Some of these are syndromic (platelet dysfunction or bone marrow failure), but others are asymptomatic. Additional germ line predisposition syndromes are likely to emerge from the broader recognition of inherited predisposition syndromes in hematologic neoplasia (eg, SAMD9/SAMD9L).27,28 With the increased recognition of germ line predisposition syndromes in myeloid cancer, the importance of accurate personal and family histories becomes apparent. Molecular profiling identifies a host of potentially germ line mutational events, and some institutions have developed expert multidisciplinary panels to identify patients with potential germ line events for whom genetic counseling and additional testing may be appropriate29 (Courtney D. DiNardo and Eric Padron, personal communication, 12 August 2018). In some series, up to 21% of patients with secondary AML will have germ line mutations in BRCA1, BRCA2, CHEK2, PALB2, or TP53.28,30 AML patients with suspected germ line events who are targeted for upfront transplantation or who relapse may benefit from additional testing (of skin fibroblasts or germ line tissue) for confirmation; this is important not only for the affected patient but for his or her family as well. Such testing is critical when considering selection of an appropriate stem-cell donor, which might require exclusion of a germ line event in a sibling.31 Our center and many others have adopted routine referral, sometimes in the hospital, for genetic counseling and testing in patients with a provoking personal or family history, especially in the context of planned allogeneic transplantation.
Good-risk disease
Among the most common genetic abnormalities in AML (occurring in ∼10% to 12% of cases) are alterations in core binding factor, which result from balanced translocations such as t(8;21) and inv(16)/t(16;16).6 These patients are generally considered to have a good prognosis, but mutations in KIT, particularly among those with t(8:21), have been suggested to adversely affect this prediction.32 More recently, molecular profiling in relatively large cohorts of uniformly treated patients with core binding factor leukemia has demonstrated frequent mutations in a spectrum of TK pathway genes [observed in up to 65% of those with t(8;21) AML and 80% of those with inv(16)] in 1 series.12,33 Of particular interest are the targetable mutations in KIT and FLT3, which were associated with higher rates of relapse, and mutations in NRAS and KRAS, which were associated with improved survival.33,34 Comutations in TK and chromatin modifier genes (ASXL1/2, EZH2, KDM6A, BCOR, and BCORL1) and/or cohesion genes (RAD21, STAG2, SMC1A, and SMC3) were identified as a particularly adverse combination more prominent in t(8;21) patients, predicting relapse rates of ∼50%.11,33 A second group recently identified the cooccurrence of >1 mutation in TK/RAS pathway genes (KIT, NRAS, KRAS, FLT3, CBL, and JAK2) to be independently associated with higher rates of relapse in core binding factor patients; these features were again more prominent in the cohort of patients with t(8;21).35 These additional molecular observations in a relatively large number of genes may in the future modify our approach to patients with putatively good-risk disease by identifying those at higher risk of relapse. Such patients might be managed differently, perhaps with a more intense consolidation approach (eg, allogeneic transplantation in first remission) and offered targeted therapy with TK inhibitors (TKIs), either off label or in the context of a clinical trial (eg, trial registered at www.clinicaltrials.gov as #NCT02013648). Some groups have adopted targeted testing for KIT and TKI therapy for patients with core binding factor leukemia, but an argument can be made for the value of a broader mutational panel covering both TKs and chromatin modifier genes, ideally in the context of a prospective study for validation of their clinical impact.
Assessment of patients with normal karyotype AML for the presence of NPM1 mutations is now a standard of care in AML. Mutations in NPM1, which occur in ∼30% of patients, are associated with a good response to induction chemotherapy, even in patients of older age.11,12,22 Within the ELN 2017 classification schema, patients with NPM1 mutations with wild-type (WT) FLT3 or with a low allelic ratio of FLT3-ITD expression are defined as good risk, but it is not just NPM1 and FLT3 interactions that matter. In the Papaemmanuil et al12 series, which included 418 patients with NPM1 mutations, the most frequently observed comutations were DNMT3A (54%), FLT3-ITD (39%), NRAS (19%), TET2 (16%), PTPN11 (15%), and IDH1/IDH2R140 (<15%). Within this cohort of relatively younger patients, cooccurrence of mutations in NPM1, FLT3-ITD, and DNMT3A predicted a particularly poor prognosis, whereas the presence of NRAS seemed to mitigate the adverse prognostic effect of FLT3-ITD. More recently, several groups including the Alliance and SWOG/UK Medical Research Council groups, have assessed the impact of NPM1 mutations in older patient cohorts, and these analyses suggest that the positive prognostic impact of NPM1 mutations may be less marked, particularly in those patients age >65 years.2,36,37
The Alliance group focused on fit older patients receiving intensive induction who did not undergo allogeneic transplantation in first remission.2 In this cohort of patients with a median age of 69 years (range, 60-85 years), 50% of whom had normal karyotype, the presence of NPM1 mutations was strongly associated with the achievement of CR (81%), but only cooccurrence of mutations in chromatin modifiers IDH2, SF1, and/or SRSF2 predicted improved overall survival (OS; 45% at 3 years).2 The absence of these coevents predicted relatively poor 3-year survival of only 11% (P < .001), and the identification of mutations in U2AF1 or WT1 were independently associated with poor outcome.2 On the basis of these data, mutational profiling with a number of genes beyond FLT3, particularly the chromatin modifier genes, splicing factor genes, IDH2, and SF1, seems to add substantially to the prognostic significance of NPM1 mutations, especially in those patients age >65 years.
The identification of biallelic mutations in CEBPA (CEBPAmut) are also considered within the good-risk paradigm by ELN 2017. These patients are in general younger, and their disease is associated with fewer cooccurring mutational events. Within the Papaemmanuil et al12 cohort, 66 patients with CEBPAmut were described, among whom 30% carried comutations with NRAS, 21% with WT1, and 20% with GATA2.12 In general, such patients have a good prognosis, but it is important to consider the family history, because like RUNX1 and TP53, CEBPA can be a germ line event.27 Indeed, 5% to 7% of patients with CEBPAmut leukemia will have a germ line predisposition,31,38 and despite an excellent rate of CR, such patients have a significant risk of disease recurrence or development of a second leukemia in another germ line clone.31
Intermediate-risk disease
The ELN defines intermediate-risk patients as those with cytogenetic abnormalities not qualified as favorable or adverse and with mutated NPM1 cooccurring with a low level FLT3-ITD (<0.5 allelic ratio) or WT NPM1 without the cooccurrence FLT3-ITD or with FLT3-ITDlow (without adverse-risk genetic lesions). Recall that comutations that occur in patients with NPM1 were addressed at length in the previous section, because this group overlaps with good-risk disease. The ELN also includes the t(9;11) (p21.3;q23.3) MLLT3-KMT2A category in this group, although within the Alliance cohort of older patients, this lesion was found to have an unfavorable prognosis.2,6 In general, patients who fall within the intermediate category present a greater challenge for the practicing clinician, because outcomes have the broadest range, and older patients with normal karyotype often fall within this cohort. Molecular profiling in this group can therefore be of particular help in choosing a course of therapy.
Of particular importance within this group is identification of patients with disease biology more like the MDSs. Such patients are characterized by mutational events within chromatin modifiers or RNA splicing genes.7,12 Within the Papaemmanuil et al12 cohort, this group was the second largest identified and included 18% of patients, 84% of whom would be classified as intermediate by the ELN. This group was defined by mutations in chromatin modifiers (ASXL1 [now poor risk], STAG2, BCOR, MLLPTD, EZH2, and PHF6) cooccurring with mutations in RNA splicing factors (SRSF2, SF3B1, U2AF1, and ZRSR2) or RUNX1 (now poor risk). Patients in this group were on average slightly older, with lower white counts and a higher rate of antecedent myeloid disorders. They had a poor response to induction therapy and a higher rate of relapse. Notably in this relatively younger cohort, 91% of patients had de novo disease and did not demonstrate morphologic dysplasia, suggesting that biologic similarity may supersede clinical history in driving outcome.39-41 A similar mutational landscape has been described in patients with high-risk MDS, secondary AML, and accelerated myeloproliferative neoplasms, all of which share a dismal prognosis with chemotherapy alone.2,7,10,11,42 Used prospectively in an otherwise intermediate-risk population, this group of mutational events might help identify those with a shared molecular pathogenesis resulting in poor outcome, who should be targeted for enrollment in clinical trials, upfront allogeneic transplantation, or early incorporation of targeted therapeutics. Several groups have shown that allogeneic transplantation can sometimes overcome adverse molecular and cytogenetic features, with the exception of TP53, for which the benefit of allografting remains controversial.43,44
Poor-risk disease
Recently added to the ELN poor-risk group are patients with mutations in TP53, RUNX1, and ASXL1 and those with a monosomal karyotype (defined as a single monosomy [except loss of X or Y] in association with at least 1 additional monosomy or structural chromosomal abnormality).6 The monosomal karyotype group was identified in the Papaemmanuil et al12 cohort as having a high preponderance of comutations in TP53 and was not mutually inclusive with complex karyotype disease. This group of patients is now well established as having a poor prognosis, with the benefits of high-dose therapy and allogeneic transplantation a topic of some debate.4,5 Although a majority of clinicians would likely offer allogeneic transplantation to these patients, long-term survival is by no means assured.43,44 The early identification of this population offers an opportunity for the adoption of novel therapeutic strategies, particularly in the context of clinical trials.45 Also included in the poor-risk category are those with secondary AML, defined as patients with prior chemotherapy exposure or an antecedent hematological diagnosis. In such patients, molecular profiling with a panel that includes genes associated with solid tumors as well as TP53 is appropriate.29,40
Unlike mutations in TP53, the adverse impact of mutations in ASXL1 and RUNX1, in combination with the use of FLT3 inhibitors for patients with high expression of FLT3-ITD (allelic ratio >0.5), seems to be at least partially overcome with the use of upfront allogeneic transplantation strategies.43,44 Appropriate early recognition of these mutations is critical to the management of newly diagnosed patients with AML who are eligible for transplantation and/or for the upfront and posttransplantation addition of TKIs or other targeted therapies.14,19,43,46
MRD for selection of postremission therapy
Recently, a plethora of publications on the use of MRD have demonstrated the power of sensitive techniques such as flow cytometry (sensitive to ∼1 per 10 000 leukemia cells) and molecular monitoring techniques for MRD assessment (sensitive to between 1 per 10 000 and 1 per 1 000 000 leukemic cells, depending on the assay used) to identify patients at high risk of relapse and to select postremission therapy. Although MRD assessment obtained after the first induction has become a standard of care in the management of acute lymphoblastic leukemia, to date its value for management of patients with AML remains unproven. Several groups have shown that patients with detectable MRD at the time of transplantation have higher rates of relapse, and some studies suggest that, at least in pediatrics, treatment choices (transplantation vs chemotherapy) might be effectively driven by MRD.14,47,48 As it stands, outside the context of a clinical trial, insufficient data exist for the routine adoption of MRD assessment in the upfront management of AML; moreover, for high-risk patients, in the absence of effective alternatives, allogeneic transplantation offers the only avenue of curative potential.
Approaches for the identification of molecular MRD include real-time polymerase chain reaction (dependent upon identification of a suitable abnormality) and next-generation sequencing (NSG; theoretically applicable to all leukemia-specific gene combinations). A majority of reports examining MRD have included younger patients, but the biological premise of such testing can logically be extended across any age category. Indeed, molecular profiling for single genes is already familiar to many of us because it is a standard of care for monitoring disease response and relapse in acute promyelocytic leukemia as well as in leukemias associated with the Philadelphia chromosome. Efforts to standardize the assessment of MRD in AML are under way, and a consensus document was recently published by the ELN Working Party.49 Although considerable variations in technique and testing methodology exist, it is clear that detection of MRD is a valuable tool for the prediction of relapse in AML (as reviewed by Paietta48 ). The ELN suggests disease profiling at the time of diagnosis, after 2 cycles of standard induction or consolidation chemotherapy, and at the end of treatment (Figure 1). It further advocates for testing in both the peripheral blood and bone marrow; the former may provide better prognostic stratification and is certainly substantially easier for the patient.49 The choice of assay used for MRD assessment is also vitally important. Most commercially available molecular assays are validated only on samples composed of >20% malignant cells and are therefore inappropriate for assessment of MRD. Use of assays designed for diagnostic assessment in this context confers no reliable information, is expensive, and might be falsely reassuring.
Mutations in NPM1, which occur in up to 30% of patients, are the most frequently identified molecular event in AML. This gene has been validated as a good marker for assessment of MRD and as a predictor for disease relapse, but the currently available tests are not uniformly validated for detection of low-level disease.14,50 Despite this, detection of MRD using NPM1 has been shown to successfully identify a subset of patients at substantial risk of relapse.50 Assay techniques for NPM1 have been laborious, but novel sequencing–based systems are likely to make MRD assessment using NPM1 more practical.15
The next most common molecular event in AML, FLT3-ITD, is present in 25% of patients. FLT3-ITD is acquired late in leukemogenesis and can be lost at the time of relapse, limiting its utility as a marker for MRD. Until recently, assays for FLT3 mutations have been unreliable because of technical issues resulting from the ITD sequence, but a recent publication suggests a novel approach to assessment of this marker.19 This assay is being validated in the context of an ongoing clinical trial and may soon be available, although given its unreliable retention at the time of relapse, its use may be most appropriate in the context of targeted therapeutics rather than true MRD monitoring.
One of the challenges of using molecular testing for MRD assessment in patients with AML is the sheer number of distinct genetic lesions in patients with AML and the large number of different mutation combinations that define disease (median of 3 mutations per patient (r) with as many as 111 different mutations (n) means 221 815 different possible combinations = n!/r!(n−r)!). Additional complexity is introduced by the presence of mutations associated with CHIP, including DNMT3A, TET2, ASXL1, or germ line events, the presence of which can persist in the context of disease clearance.13,15,16,18 Recently, the Dutch group published results from a combined MRD assessment using targeted NGS for a 54-gene panel with flow cytometry. Patients in this study had a diagnosis of AML (428 patients) or high-risk MDS (54 patients) and had achieved remission (<5% bone marrow blasts) after 2 cycles of induction. The primary and secondary end points were 4-year cumulative incidence of relapse, OS, and relapse-free survival. The authors identified an average of 3 mutations per patient, with at least 1 marker identified in 89% of patients (430 of 482). In this cohort, isolated persistence of mutations in the CHIP genes was not associated with relapse. By contrast, detection of other mutations at any level, and at any time point between day 21 and 4 months after induction (observed in 28% of patients), strongly predicted relapse (55% vs 32%; P < .001), relapse-free survival (58% vs 37%; P < .001), and OS (66% vs 42%; P < .001) and remained significant in a multivariable analysis including established prognostic factors (age, white blood cell count, ELN 2017, number of cycles to CR). Notably, in this cohort, flow cytometric assessment of MRD conferred independent prognostic value and, combined with molecular MRD, predicted relapse risk in 73% of patients, suggesting that each modality provides nonoverlapping information. Allogeneic transplantation was also found to be protective for relapse (hazard ratio, 0.45; P < .001) but not OS. Additional groups have reported on the use of molecular MRD and have demonstrated similar findings.19,47,51
The takeaway from these data is that to be applicable to a majority of AML patients, MRD panels must cover a broad range of genes and be validated at low disease burden, and detection of the CHIP genes must be discounted. Furthermore, MRD assessment strategies should probably include assessment of MRD by flow cytometry (although which method to use remains controversial).49 Emphasizing the importance of these data for prognosis, the ELN Working Party has proposed MRD− CR as a new therapeutic end point for patient management, and several clinical trials are in progress to validate this end point (Table 2).6 Sadly, even the best combined approach fails to predict relapse in 25% to 30% of patients.
Ongoing interventional trials using MRD-directed therapies
| Sponsor/trial | Identifier | Phase | Estimated N of participants | Patient age, y | Clinical notes | MRD assessment | MRD+ intervention | Expected completion |
|---|---|---|---|---|---|---|---|---|
| Cardiff University; UK MRC AML 18 | NCT02272478 | 3 | 1600 | >60 | Older patients with primary or secondary AML or high-risk MDS; DA or DA + GO induction | Postinduction, flow cytometry based | MRD+ patients randomized to DA, DA + cladaribine, or FLAG-Ida | October 2020 |
| Hospital La Fe, Spain; PETHEMA-LMA 10 | NCT01296178 | 3 | 200 | <65 | Primary or secondary, treatment-naïve, non-APL AML patients; cytarabine + Ida induction | Flow cytometry and molecular based | Risk-adapted, MRD-directed therapy, not specified | December 2018 |
| Various sites in Germany; RELAZA2 | NCT01462578 | 2 | 93 | >18 | AML or MDS patients with detectable molecular markers after chemotherapy or allo-SCT | Patient-specific molecular markers [eg, mutant NPM1, t(6,9), or decrease in CD34 chimerism] | 6-12 additional cycles of azacitidine | June 2020 |
| MD Anderson Cancer Center, Houston, TX | NCT02684162 | 2 | 90 | 15-75 | Patients with AML, MDS, or CMML and posttransplantation relapse | Flow cytometry, cytogenetics, or molecular markers | SGI-110 + DLI | June 2022 |
| University Chicago, Chicago, IL, and various sites in United States | NCT02275533 | 2 | 80 | >18 | Patients in CR1 or CRi after induction or consolidation | Longitudinal PCR for WT1 expression | Randomized to nivolumab vs observation | June 2019 |
| University California San Francisco, San Francisco, CA | NCT02458235 | 2 | 67 | <30 | AML, ALL, MDS, or JMML patients undergoing allo-SCT; risk determined by GVHD, chimerism, and MRD status | Flow cytometry + gene expression profiling when feasible | Azacitidine + DLI in high-risk patients | December 2019 |
| Institute of Hematology and Blood Diseases Hospital, Tianhin, China | NCT03021395 | 1/2 | 300 | 14-55 | Primary AML in CR, postconsolidation | Postconsolidation, not specified | Decitabine for patients not undergoing transplantation | December 2022 |
| Sponsor/trial | Identifier | Phase | Estimated N of participants | Patient age, y | Clinical notes | MRD assessment | MRD+ intervention | Expected completion |
|---|---|---|---|---|---|---|---|---|
| Cardiff University; UK MRC AML 18 | NCT02272478 | 3 | 1600 | >60 | Older patients with primary or secondary AML or high-risk MDS; DA or DA + GO induction | Postinduction, flow cytometry based | MRD+ patients randomized to DA, DA + cladaribine, or FLAG-Ida | October 2020 |
| Hospital La Fe, Spain; PETHEMA-LMA 10 | NCT01296178 | 3 | 200 | <65 | Primary or secondary, treatment-naïve, non-APL AML patients; cytarabine + Ida induction | Flow cytometry and molecular based | Risk-adapted, MRD-directed therapy, not specified | December 2018 |
| Various sites in Germany; RELAZA2 | NCT01462578 | 2 | 93 | >18 | AML or MDS patients with detectable molecular markers after chemotherapy or allo-SCT | Patient-specific molecular markers [eg, mutant NPM1, t(6,9), or decrease in CD34 chimerism] | 6-12 additional cycles of azacitidine | June 2020 |
| MD Anderson Cancer Center, Houston, TX | NCT02684162 | 2 | 90 | 15-75 | Patients with AML, MDS, or CMML and posttransplantation relapse | Flow cytometry, cytogenetics, or molecular markers | SGI-110 + DLI | June 2022 |
| University Chicago, Chicago, IL, and various sites in United States | NCT02275533 | 2 | 80 | >18 | Patients in CR1 or CRi after induction or consolidation | Longitudinal PCR for WT1 expression | Randomized to nivolumab vs observation | June 2019 |
| University California San Francisco, San Francisco, CA | NCT02458235 | 2 | 67 | <30 | AML, ALL, MDS, or JMML patients undergoing allo-SCT; risk determined by GVHD, chimerism, and MRD status | Flow cytometry + gene expression profiling when feasible | Azacitidine + DLI in high-risk patients | December 2019 |
| Institute of Hematology and Blood Diseases Hospital, Tianhin, China | NCT03021395 | 1/2 | 300 | 14-55 | Primary AML in CR, postconsolidation | Postconsolidation, not specified | Decitabine for patients not undergoing transplantation | December 2022 |
An overview of ongoing trials that depend on MRD assessment for guidance in subsequent patient management. Observational protocols, or those where MRD data are collected but not used in treatment decisions, are excluded. Trials focused solely on pediatric populations are also excluded.
ALL, acute lymphoblastic leukemia; allo-SCT, allogeneic stem-cell transplantation; APL, acute promyelocytic leukemia; CMML, chronic myelomonocytic leukemia; CR1, first CR; CRi, incomplete CR; DA, daunorubicin plus cytarabine; DLI, donor lymphocyte infusion; FLAG, fludarabine, cytarabine, and granulocyte colony-stimulating factor; GO, gemtuzumab ozogamicin; GVHD, grant-versus-host disease; Ida, idarubicin; JMML, juvenile myelomonocytic leukemia.
IDH1/2 mutations: target and MRD
Somatic mutations in genes encoding citric acid cycle enzymes isocitrate dehydrogenase 1 (IDH1) and IDH2 are present in up to 20% of AML cases.52 A trio of recurrent IDH mutations at IDH1-R132, IDH2-R172, and IDH2-R140 promote the disabling of α-ketoglutarate–dependent enzymes and the formation of R-2-hydroxyglutarate.46 This oncometabolite drives leukemogenesis by disrupting cellular differentiation programs and TET2-mediated DNA methylation. A landmark 2017 phase 1/2 clinical trial led to the US Food and Drug Administration approval of enasidenib for the treatment of relapsed/refractory IDH2-mutant disease.53 A companion diagnostic polymerase chain reaction assay was also approved. More recently, work by DiNardo et al54 led to the approval of a second targeted agent for IDH1-mutated AML. Given the availability of effective therapies targeting IDH1/2, mutational assessment of these genes has been widely adopted in the frontline and relapsed/refractory settings, but the clinical implication of these events remains unconfirmed. Some groups have shown no significant impact on outcome,52 whereas other investigators have found an adverse impact (Patel BJ, Thota S, Carraway H, Griffiths EA, Thompson JE, Scully M, Kuzmanovic T, Barot S, Hirsch C, Prahxhjhj B, Przychodzen B, Nazha A, Mukherjee S, Gerds A, Sekeres M, Kalaycio M, Advani A, Wang ES, Meggendorfer M, Maciejewski JP, manuscript submitted). At the time of publication of the 2017 recommendations, the ELN did not feel there was sufficient evidence to assign IDH1- or IDH2-mutated AML to a formal risk category (mutations in DNMT3A and chromatin modifier/spliceosome genes also did not meet the bar for inclusion; Table 1).
Although the exact prognostic influences of IDH mutations remain controversial, experts generally feel that IDH1- or IDH2-mutant disease corresponds to intermediate-risk AML, frequently clustering with NPM1 mutations and intermediate-risk cytogenetics.52 IDH-mutated patients tend to be older and have higher blast percentages and are less likely to have secondary disease compared with IDHWT AML patients.
The use of IDH1/2 mutations alone as markers of MRD is generally discouraged49 because of their clonal instability over time and their contribution to clonal hematopoiesis.50 Longitudinal IDH mutational testing for biomarker purposes would not be helpful, because VAFs of IDH mutations detected at diagnosis have been shown to remain steady during initial disease progression. However, as IDH-mutant–containing cells that survive chemotherapy spawn subclones, situations can arise where the emergence of new mutants not detected at diagnosis predominate at relapse.46 Hematologists who seek to use molecular tests designed to monitor treatment of IDH1/2-mutant AML should remain mindful that enasidenib and ivosidenib exert therapeutic activity by causing mutant cells to differentiate rather than to die, and as a result, VAF kinetics may not follow the same pattern as other molecular events.55
Mutations and relapsed/refractory AML
Since the sequencing of the first AML genome in 2008, AML has been recognized as a complex and dynamic disease associated with multiple somatic mutational events and clonal evolution in the context of therapy.56-58 For patients with relapsed or refractory AML, mutational profiling is most useful to identify potentially targetable mutational events amenable to treatment in clinical trials or with approved therapies. Reassessment of the mutational profile at the time of relapse is necessary because clonal evolution can result in loss of some targets, with expansion of subclones present at low levels at the time of original disease assessment (Figure 1). Inhibitors of protein kinases, epigenetic modulators, splicing inhibitors, novel immunotherapies, and drugs targeting oncogenic proteins and the microenvironment are in development, and many of these programs have targeted specific subsets of genetically defined AML for enrichment. Therapeutic interventions targeting founding mutations in epigenetic modifier genes are hypothesized to more effectively target leukemia-initiating cells, and inhibitors of TKs like FLT3 and RAS are thought to target leukemic subclones; therefore, combination strategies are likely required, particularly in relapsed/refractory disease. Strategies aimed at bringing such approaches early (eg, the BEAT AML trial; #NCT02927106) are designed to maximize efficiency in identifying synergistic therapies based upon mutational profiling, and we may be hopeful that this strategy will produce lower rates of relapse and refractory disease.
Conclusions
Approved drugs targeting mutational events in FLT3, JAK/STAT signaling, IDH1, and IDH2 are already on the market. Clinical trials of drugs targeting RNA splicing and a myriad of other pathways exposed by mutational analysis are under way, spurred by the widespread adoption of molecular profiling in patients with newly diagnosed and relapsed/refractory AML and other myeloid cancers. The approval of new drugs, the upfront application of which is driven by a mutational as well as prognostic risk profile (eg, midostaurin for FLT3-mutant AML, liposomal daunorubicin/cytarabine for AML with MDS-related changes or secondary disease, and gemtuzumab ozogomycin, the beneficial impact of which seems most pronounced in patients with good- or intermediate-risk disease), forces us to appropriately assess and stratify our patients with increasing speed. It is clear that patients with newly diagnosed AML should undergo mutational profiling for at least the ELN 2017–recommended genes, but it is also clear that distinct cytogenetic and molecular comutational events are important modifiers of response to standard therapy (Table 1). Given the decreasing cost of NGS approaches, which allow assessment of a large number of genes, it seems rational to cast a broad net. Gene panel sequencing approaches that include some minimum number of relatively frequent, potentially targetable, and disease-defining mutational events as well as germ line mutations will contribute to our approach to upfront management and identify patients within distinct biological subgroups. This approach is likely to help identify patients who will respond best to particular therapies and improve our fundamental understanding of this complex disease.
If validated, assessment of MRD at the time of established remission will allow modification of our therapeutic approach to offer upfront allogeneic transplantation for those most likely to benefit, while sparing those without MRD this toxic and life-altering approach. Several groups are actively pursuing such MRD-based strategies (#NCT01452646; Table 2). Broad availability of novel sequencing techniques are necessary to distinguish those patients at high risk of relapse and therefore appropriate for allogeneic transplantation or clinical trials of novel therapy from those more likely to remain in remission, who require less intensive treatment (#NCT02275533). Availability of such an assessment is likely to change clinical practice.
Correspondence
Elizabeth A. Griffiths, Roswell Park Comprehensive Cancer Center, Elm & Carlton Sts, Buffalo, NY 14263; e-mail: elizabeth.griffiths@roswellpark.org.
This article was selected by the Blood Advances and Hematology 2018 American Society of Hematology Education Program editors for concurrent submission to Blood Advances and Hematology 2018. It is reprinted from Blood Advances 2018, Volume 2.
Off-label drug use: none disclosed.
