

Abstract
Both older and newer cell therapies have demonstrated impressive responses in otherwise poor-prognosis lymphomas. Consequently, cellular therapy now plays a major role in the management of many non-Hodgkin lymphomas. In this article, we examine the role of chimeric antigen receptor (CAR) T cells, allogeneic stem cell transplantation, and virus-directed T cells for treatment of lymphomas. We review the current indications for CAR T cells and discuss our clinical approach to selecting and treating patients with aggressive B-cell lymphomas to receive CD19-directed CAR T cells. In addition, we highlight newer cell therapies and provide an overview of promising future approaches that have the potential to transform immunotherapy with cells to treat lymphomas.
Learning Objectives
Describe the role of CAR T cells in the treatment of lymphoma
Identify limitations to currently available CAR T-cell therapies
Review the role of allogeneic stem cell transplantation in B-cell lymphomas
Discuss novel approaches to cell therapies
Clinical case 1
A 50-year-old woman with diffuse large B-cell lymphoma (DLBCL) initially received 6 cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) chemotherapy but relapsed within 3 months. She then received 2 cycles of R-ICE (rituximab, ifosfamide, carboplatin, and etoposide), and positron emission tomography/computed tomography (PET/CT) showed stable disease. After discussing therapeutic options, including alternative chemotherapy vs chimeric antigen receptor (CAR) T cells (CAR-T), she proceeded with CAR-T. After leukapheresis for T-cell collection, she presented with rapidly enlarging lymphadenopathy. Bridging therapy with polatuzumab-bendamustine/rituximab was initiated, and, after 2 cycles, PET/CT showed an excellent partial response and a normalized level of lactate dehydrogenase (LDH). She underwent lymphodepletion with cyclophosphamide/fludarabine followed by infusion of tisagenlecleucel.
This case raises the following questions: which patients should be referred for consideration of cellular therapy, when should therapy be initiated, and what should the management considerations be for patients undergoing CAR-T?
CD19-directed CAR T cells
Aggressive B-cell lymphomas: US Food and Drug Administration−approved products
CD19-directed CAR-T is an option for patients with DLBCL, high-grade B-cell lymphoma (HGBCL), transformed follicular lymphoma (tFL), and primary mediastinal large B-cell lymphoma (PMBL) that is relapsed/refractory after 2 or more lines of therapy.
The SCHOLAR-1 study provided a benchmark for the very poor outcomes in patients with refractory DLBCL before the availability of CAR-T. Median overall survival (OS) was 6.3 months, and only 20% of patients remained alive at 2 years.1
The currently approved CAR-T products improve on these historical outcomes. CARs are synthetic molecules containing an extracellular single-chain variable fragment directed against a tumor antigen such as CD19, as well as a hinge region, a transmembrane domain, and an intracellular signaling domain. CAR-Ts are manufactured from T-cells and are genetically modified to express the CAR on the cell surface.2 The “first-generation” CAR-Ts contained the CD3ζ signaling domain and had limited expansion, persistence, and antitumor activity. A major breakthrough came with the addition of a costimulatory domain, such as 4-1BB or CD28, to the CAR molecule, resulting in dramatic improvement in expansion, persistence, and T-cell killing. CAR-Ts recognize their target in a major histocompatibility class–unrestricted manner and activate the T-cell signaling and costimulatory pathways. The CARs best studied in lymphoma are diagrammed in the accompanying visual abstract; Table 1 summarizes the properties of these CAR-Ts.
CD19-directed CAR T-cell products for DLBCL: 1-year outcomes
| Axicabtagene ciloleucel ZUMA-1 trial3,4 | Tisagenlecleucel JULIET trial5,6 | Lisocabtagene maraleucel TRANSCEND NHL 001 trial10 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| US FDA approved | Yes | Yes | No | |||||||
| CAR construct | Anti-CD19, CD28, CD3z | Anti-CD19, 4-1BB, CD3z | Anti-CD19, 4-1BB, CD3z (tEGFR) | |||||||
| Costimulatory domain | CD28 | 4-1BB | 4-1BB | |||||||
| Vector | Retrovirus | Lentivirus | Lentivirus | |||||||
| CAR T-cell manufacturing | Bulk, fresh | Bulk, cryopreserved | CD8+ and CD4+ T cells: separate, fresh | |||||||
| CAR T-cell dose | 2.0 × 106 cells/kg, max 2.0 × 108 cells | 0.6-6 × 108 cells | 1.0 × 108 CD8+ and CD4+ cells | |||||||
| Bridging therapy | No | Yes: 92% | Yes: 59% | |||||||
| Lymphodepletion | Flu/Cy (30 mg/m2, 500 mg/m2) × 3 d | Flu/Cy (25 mg/m2, 250 mg/m2) × 3 d or bendamustine (90 mg/m2) × 2 d | Flu/Cy (30 mg/m2, 300 mg/m2) × 3 d | |||||||
| Secondary CNS lymphoma | No | No | Yes: small number | |||||||
| ALC cutoff for manufacturing, per µL | ALC ≥100 | ALC ≥300 | None | |||||||
| Lymphoma subtypes enrolled | DLBCL/HGBCL | PMBL | tFL | DLBCL/ HGBCL | tFL | DLBCL | HGBCL | t-iNHL | PMBL | FL3B |
| Evaluable patients, n | 77 | 8 | 16 | 89 | 22 | 137 | 36 | 78 | 15 | 3 |
| Follow-up time, mo | 15.4 | 14 | 12.3 | |||||||
| Efficacy, n | 101 | 93 | 256 | |||||||
| Best ORR, % (CR%) | 82 (54) | 52 (40) | 73 (53) | |||||||
| DOR at 12 mo | 11.1 mo/NR* | NR | NR (all patients) | |||||||
| 5.6 mo | 10.8 mo | NR (tFL) | NR | — | ||||||
| DOR for CR at 12 mo | NR | NR | NR | |||||||
| OS at 12 mo, % | 59 | 49 | 58 | |||||||
| Median follow-up for trial, mo | 27 | 24 | 12 | |||||||
| Safety, n | 101 | 111 | 269 | |||||||
| CRS ≥grade 3, % | 13† | 22‡ | 2‡ | |||||||
| CRS time to onset median duration (range) | 2 d (range, 1-12) | 3 d (range, 1-9) | 5 d (range, 1-14) | |||||||
| 8 d (not reported) | 7 d (range, 2-30) | 5 d (1-17) | ||||||||
| Neurotoxicity ≥grade 3, % | 28 | 12 | 10 | |||||||
| Neurotoxicity time to onset median duration (range) | 5 d (range, 1-17) | 6 d (range, 1-17) | 9 d (range 1-66) | |||||||
| not reported | 14 d (not reported) | 11 d (range, 1-86) | ||||||||
| Axicabtagene ciloleucel ZUMA-1 trial3,4 | Tisagenlecleucel JULIET trial5,6 | Lisocabtagene maraleucel TRANSCEND NHL 001 trial10 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| US FDA approved | Yes | Yes | No | |||||||
| CAR construct | Anti-CD19, CD28, CD3z | Anti-CD19, 4-1BB, CD3z | Anti-CD19, 4-1BB, CD3z (tEGFR) | |||||||
| Costimulatory domain | CD28 | 4-1BB | 4-1BB | |||||||
| Vector | Retrovirus | Lentivirus | Lentivirus | |||||||
| CAR T-cell manufacturing | Bulk, fresh | Bulk, cryopreserved | CD8+ and CD4+ T cells: separate, fresh | |||||||
| CAR T-cell dose | 2.0 × 106 cells/kg, max 2.0 × 108 cells | 0.6-6 × 108 cells | 1.0 × 108 CD8+ and CD4+ cells | |||||||
| Bridging therapy | No | Yes: 92% | Yes: 59% | |||||||
| Lymphodepletion | Flu/Cy (30 mg/m2, 500 mg/m2) × 3 d | Flu/Cy (25 mg/m2, 250 mg/m2) × 3 d or bendamustine (90 mg/m2) × 2 d | Flu/Cy (30 mg/m2, 300 mg/m2) × 3 d | |||||||
| Secondary CNS lymphoma | No | No | Yes: small number | |||||||
| ALC cutoff for manufacturing, per µL | ALC ≥100 | ALC ≥300 | None | |||||||
| Lymphoma subtypes enrolled | DLBCL/HGBCL | PMBL | tFL | DLBCL/ HGBCL | tFL | DLBCL | HGBCL | t-iNHL | PMBL | FL3B |
| Evaluable patients, n | 77 | 8 | 16 | 89 | 22 | 137 | 36 | 78 | 15 | 3 |
| Follow-up time, mo | 15.4 | 14 | 12.3 | |||||||
| Efficacy, n | 101 | 93 | 256 | |||||||
| Best ORR, % (CR%) | 82 (54) | 52 (40) | 73 (53) | |||||||
| DOR at 12 mo | 11.1 mo/NR* | NR | NR (all patients) | |||||||
| 5.6 mo | 10.8 mo | NR (tFL) | NR | — | ||||||
| DOR for CR at 12 mo | NR | NR | NR | |||||||
| OS at 12 mo, % | 59 | 49 | 58 | |||||||
| Median follow-up for trial, mo | 27 | 24 | 12 | |||||||
| Safety, n | 101 | 111 | 269 | |||||||
| CRS ≥grade 3, % | 13† | 22‡ | 2‡ | |||||||
| CRS time to onset median duration (range) | 2 d (range, 1-12) | 3 d (range, 1-9) | 5 d (range, 1-14) | |||||||
| 8 d (not reported) | 7 d (range, 2-30) | 5 d (1-17) | ||||||||
| Neurotoxicity ≥grade 3, % | 28 | 12 | 10 | |||||||
| Neurotoxicity time to onset median duration (range) | 5 d (range, 1-17) | 6 d (range, 1-17) | 9 d (range 1-66) | |||||||
| not reported | 14 d (not reported) | 11 d (range, 1-86) | ||||||||
Axicabtagene ciloleucel (axi-cel) is a CD28-containing CAR-T and has been studied for relapsed/refractory DLBCL, tFL, PMBL, and HGBCL. The complete remission (CR) rate was 59%, median duration of response (DOR) was 11.1 months, and 2-year OS was 50.5%.3,4 Tisagenlecleucel (tisa-cel) is a 4-1BB–containing CAR-T and has been studied for relapsed/refractory DLBCL, tFL, and HGBCL. After tisa-cel, 40% of patients achieved CR, median DOR was not reached, and OS was 10.3 months (Table 1).5,6
Responses to CAR-Ts are generally rapid and occur within 1 to 3 months. For patients in remission beyond 6 to 12 months, most of the responses remain durable, as noted at 3- and 4-year follow-ups.7-9 Taken together, the results suggest that at least some patients may be cured of lymphoma after CAR-T.
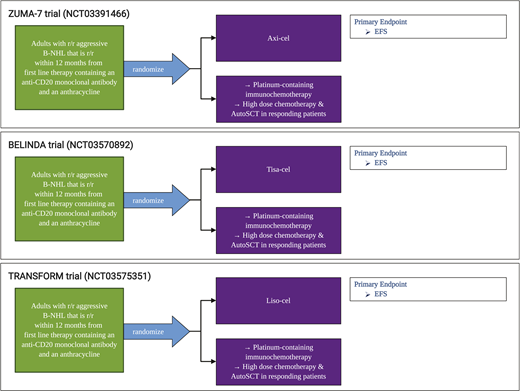
In our practice, patients with aggressive B-cell lymphoma are considered for anti-CD19 CAR-T when they have stable or progressive lymphoma after second-line chemotherapy, have a relapse after autologous stem cell transplantation (SCT), or require a third-line or greater therapy. Currently, patients who achieve CR and select patients with a partial response after second-line chemotherapy are offered autologous SCT. There are ongoing randomized trials comparing the role of CD19-directed CAR-T vs autologous SCT for second-line therapy (Figure 1), as well as first-line CAR-T trials for high-risk patients (www.clinicaltrials.gov #NCT03761056).
Patient selection
An important aspect of CAR-T is appropriate patient selection and management before the cells are infused. Aside from active infection at the time of CAR-T infusion, there is no absolute contraindication to CAR-T. Performance status and organ function are major considerations. Although clinical trials have enrolled only relatively fit patients (Eastern Cooperative Oncology Group performance status 0-1), real world data substantiates that patients with higher performance status can be treated safely,10 as is also our practice. If the patient is fit, there is no age at which we deem the patient ineligible for CAR-T. Both clinical trial and real-world data have failed to demonstrate an age cutoff for those who benefit from CAR-T.3,4,10,11 Nevertheless, cytokine release syndrome (CRS) and neurologic toxicity appear to increase with CD28-containing CAR-Ts, and we often select a CAR-T product with less toxicity in older or frail patients (Table 1). Because CRS can result in physiologic stress, including high fevers, capillary leak, and hemodynamic instability, patients must have adequate organ function reserve to tolerate potentially severe CRS. Clinical trials and real-world practice typically exclude patients with significant cardiomyopathy (New York Heart Association grades 3 and 4 or left ventricle ejection fraction <40%-45%), renal dysfunction (creatinine <1.6-2 mg/dL or creatinine clearance <40), liver disease, or poor lung function. Furthermore, given potential neurologic toxicity with all CAR-T products, we are hesitant to administer cells to patients with underlying cognitive impairment related to difficulty in monitoring for neurotoxicity. Responses do not seem to be affected by cell of origin or double-hit lymphoma status.3,5 However, patients with uncontrolled lymphoma, reflected by tumor volume,6,12 may have inferior outcomes. Data suggest that patients with poor performance status and elevated LDH levels also have shorter PFS and OS.10 These patients represent a high-risk group that should be addressed in future trials.
Although commercial CAR-T is not approved for primary central nervous system (CNS) lymphoma, small subsets of patients with secondary CNS lymphoma have been treated.10,11,13 Currently, there is no evidence that these patients have a higher incidence of CNS toxicity.
Another consideration is selection of therapy for patients who require disease control. Before T-cell collection, the intent is to preserve an intact immune system, especially the number of lymphocytes. We try to avoid therapies that deplete lymphocytes, including radiation, bendamustine, corticosteroids, or cytotoxic chemotherapy, immediately before collecting a patient’s T-cells for CAR-T manufacturing.
After leukapheresis, bridging therapy may be necessary to maintain control of aggressive lymphoma during commercial CAR-T manufacturing. The foremost goal of bridging therapy is to maintain functional status and organ function. Compared with therapy before T-cell collection, the choices for bridging therapy are less limited because the T-cells needed for manufacturing have already been removed from the patient. Bridging therapy can be individualized. In the current case we chose polatuzumab-bendamustine/rituximab; other options include corticosteroids, chemotherapy, radiation, monoclonal antibodies, and immunomodulatory agents. Another interesting approach that we have used for bridging therapy is ibrutinib, which is active in nongerminal center DLBCL and may also improve the number and function of T-cells.14 Some data suggest that patients who receive bridging therapy while awaiting axi-cel have an outcome inferior to that of patients who do not receive bridging therapy.10,15,16 Although bridging therapy may have a negative impact, it is more likely that this effect identifies a higher risk group of patients based on the need for disease control after T-cell collection. Theoretically, it is possible that immunosuppressive bridging therapy can enhance CAR-T expansion and persistence.
Post-CAR T-cell toxicity
The 2 well-described therapy-specific toxicities after CAR-T are CRS and neurologic toxicity (immune effector cell-associated neurotoxicity [ICANS]). The diagnosis of CRS requires the presence of fever. More severe cases progress to capillary leak and hypotension. Grading is dependent on presence and severity of hypotension and hypoxia.17 Nevertheless, manifestations can affect almost any organ.18 ICANS often manifests with headaches and various degrees of encephalopathy that adversely affect speech, level of consciousness, and motor function and can progress to seizures and cerebral edema.17
Known risk factors for CRS and ICANS include elevated LDH and high tumor burden before CAR-T infusion.6,10 Management of CRS and ICANS is now more consistently defined by consensus American Society for Transplantation and Cellular Therapy guidelines.17,19 Tocilizumab, an anti-IL6 receptor antibody, remains the mainstay of therapy for CRS. Corticosteroids are the recommended treatment for ICANS, although treatment approaches for neurologic complications are less well defined. Mitigation of CRS and ICANS is an ongoing field of study. Approaches include prophylactic or early steroids20 and tocilizumab,21,22 anakinra, and itacitinib.
Other toxicities after CAR-T infusion are cytopenias, infection, B-cell aplasia, and hypogammaglobulinemia. Patients in long-term remission may require intravenous immune globulin, although many patients have B-cell and immunoglobulin recovery.4,23
Clinical case 1 (continued)
The patient in our case was at lower risk of complications because she had a normal LDH level and decreased tumor volume after bridging therapy. She developed grade 1 CRS that spontaneously resolved. Her 3-month PET/CT showed complete remission and she continued in remission 55 months after CAR-T. Her only long-term complication was hypogammaglobulinemia. Per our institutional practice, she did not require intravenous immune globulin for the condition, because she did not have recurrent sinopulmonary infections.
Mantle cell lymphoma: FDA-approved product
Brexucabtagene autoleucel (KTE-X19) has been approved by the US Food and Drug administration (FDA) for relapsed/refractory MCL. Sixty patients previously treated with Bruton tyrosine kinase (BTK) inhibitors, anti-CD20 antibodies, and chemotherapy had an overall response rate (ORR) of 93%, a CR rate of 67%, and 1-year progression free-survival (PFS) of 61%, although longer-term follow-up is needed.24 Toxicity was similar to that reported for axi-cel (CRS, 91%; neurologic toxicity, 63%). CAR-T appeared to be efficacious regardless of historically poor-risk groups, including those with blastoid MCL.
Newer agents and non-FDA–approved indications
Lisocabtagene maraleucel for aggressive B-cell lymphomas
In addition to axi-cel and tisa-cel, another important anti-CD19 CAR-T product is lisocabtagene maraleucel (liso-cel; JCAR017). Similar to tisa-cel, this product uses a 4-1-BB costimulatory domain and lentivirus transfection. The major difference is that equal doses of CD8+ and CD4+ CAR-Ts are infused sequentially. In a trial for relapsed/refractory aggressive B-cell lymphomas, the CR rate was 53%, median PFS was 12.3 months, and OS was 58%.11 This product is currently not approved for this indication; however, the data are promising, and liso-cel may join the products that are currently commercially available for aggressive B-cell lymphomas.
In Table 1, we present a summary of the 3 CAR-T products that are furthest along in development for relapsed/refractory lymphoma. However, we stress that it is not possible to directly compare outcomes across these clinical trials. Trial design, patient characteristics and enrollment, and even toxicity grading differ across studies. Generally, responses are impressive and relatively similar for a group of patients with highly refractory lymphoma and an otherwise dismal prognosis. Toxicity varies across studies but there is low treatment–related mortality.
CD19-directed CAR T cells for chronic lymphocytic leukemia
Although CAR-T is not currently FDA approved for chronic lymphocytic leukemia (CLL), relapsed/refractory CLL was one of the first diseases treated with it. We tested anti-CD19 CAR-Ts (ultimately developed as tisa-cel) in 14 heavily pretreated patients who had relapsed/refractory CLL. Responses were durable in most patients; the first 2 patients treated remained in remission beyond 10 years.25 Liso-cel also showed promising early results for CLL.26 To further improve CAR-T efficacy, ibrutinib has been combined with CAR-T for CLL27,28 based on data that ibrutinib enhances CAR-T function in CLL.14 Optimization of CAR-T dosing may be another strategy to improve outcomes.29
CD19-directed CAR T cells for FL and marginal zone lymphoma
Studies have demonstrated high response rates8,9,23,30 and long-term remissions8,9,23 after CAR-T administration for FL (Table 2). In 80 patients with FL and 14 patients with marginal zone lymphoma that relapsed after 2 prior therapies, axi-cel treatment produced a 94% ORR with CR of 80%.30 For the tisa-cel construct, similar FL responses were observed (80% ORR) with 60% of patients responding at 4 years.8,23 Taken together, the limited long-term data suggest that remissions after anti-CD19 CAR-T in indolent lymphoma are durable.
Selected CD19-directed CAR T-cell trials
| Trial registration no. and name | Lymphoma status | N, Lymphoma subtypes | Ongoing? | ORR, % | PFS | DOR,% | Median follow-up, mo |
|---|---|---|---|---|---|---|---|
| NCT00924326 | R/r | N = 43 | No | — | EFS: | 3 y DOR: | 42 |
| NCI trial (axi-cel construct)9 | DLBCL, 28; low-grade B-NHL, 8; | DLBCL, 15 mo; | DLBCL+PMBL, 48; | ||||
| CLL, 7 | Low grade-B NHL, 55 mo; | Low-grade B-NHL, 63; | |||||
| CLL, 40.5 mo | CLL, 50 | ||||||
| NCT02030834 | No curative treatment options, <2-y life expectancy | N = 38 | No | DLBCL, 50; | DLBCL, 5.8 mo; | 49 mo DOR: | 49 |
| Penn trial (tisa-cel construct)8,23 | DLBCL, 24; | FL, 79 | FL, 32.4 mo | DLBCL, 60; | |||
| 14 FL, 14 | FL, 60 | ||||||
| NCT02601313 | R/r after BTKi, chemotherapy, and anti-CD20 antibody | N = 60 | No | 85 | 61% | Not reached | 12.3 |
| ZUMA-224 | MCL (primary analysis) | ||||||
| NCT03105336 | R/r after 2 therapies, including anti-CD20 and alkylating agent | N = 94 | Yes | 94 | — | — | — |
| ZUMA-530 | FL, 80; | ||||||
| MZL, 14 | |||||||
| NCT03331198 | R/r after 3 therapies if standard risk; r/r after 2 therapies if high risk; all received prior ibrutinib | N = 23 | Yes | 82 | — | — | — |
| TRANSCEND CLL 00426 | CLL |
| Trial registration no. and name | Lymphoma status | N, Lymphoma subtypes | Ongoing? | ORR, % | PFS | DOR,% | Median follow-up, mo |
|---|---|---|---|---|---|---|---|
| NCT00924326 | R/r | N = 43 | No | — | EFS: | 3 y DOR: | 42 |
| NCI trial (axi-cel construct)9 | DLBCL, 28; low-grade B-NHL, 8; | DLBCL, 15 mo; | DLBCL+PMBL, 48; | ||||
| CLL, 7 | Low grade-B NHL, 55 mo; | Low-grade B-NHL, 63; | |||||
| CLL, 40.5 mo | CLL, 50 | ||||||
| NCT02030834 | No curative treatment options, <2-y life expectancy | N = 38 | No | DLBCL, 50; | DLBCL, 5.8 mo; | 49 mo DOR: | 49 |
| Penn trial (tisa-cel construct)8,23 | DLBCL, 24; | FL, 79 | FL, 32.4 mo | DLBCL, 60; | |||
| 14 FL, 14 | FL, 60 | ||||||
| NCT02601313 | R/r after BTKi, chemotherapy, and anti-CD20 antibody | N = 60 | No | 85 | 61% | Not reached | 12.3 |
| ZUMA-224 | MCL (primary analysis) | ||||||
| NCT03105336 | R/r after 2 therapies, including anti-CD20 and alkylating agent | N = 94 | Yes | 94 | — | — | — |
| ZUMA-530 | FL, 80; | ||||||
| MZL, 14 | |||||||
| NCT03331198 | R/r after 3 therapies if standard risk; r/r after 2 therapies if high risk; all received prior ibrutinib | N = 23 | Yes | 82 | — | — | — |
| TRANSCEND CLL 00426 | CLL |
BTKi, BTK inhibitor; r/r, relapsed/refractory.
One challenge in the application of CAR-T to FL and other indolent lymphomas will be determining the appropriate sequencing for therapy, given the large array of therapeutic options. CAR-T has a higher early toxicity profile than most other available noncurative therapies, but preliminarily appear to induce long-term remission.
EBV-directed cytotoxic T cells
Autologous LMP1/LMP2-directed cytotoxic T lymphocytes (CTLs) have been shown to induce durable remissions in Epstein-Barr virus (EBV)–associated lymphomas.31 Tabelecleucel is an allogeneic T-cell product that utilizes donor-derived EBV-specific CTLs to treat EBV+ post-transplant lymphoproliferative disorder. HLA restriction and matching are used to select a donor product for a given patient from a preexisting library. This therapy depends on recognition of EBV by the donor T-cell receptor, which differs from the chimeric receptor used in CAR-Ts. Posttreatment follow-ups are currently short, but early results are encouraging.32,33 A multicenter phase 3 trial (NCT03394365) is under way.
Clinical case 2
A 56-year-old woman with HGBCL was treated with R-EPOCH (rituximab, etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin) but relapsed 6 months after concluding chemoimmunotherapy. After R-DHAP (rituximab, dexamethasone, cytarabine, cisplatin) followed by autologous SCT, she achieved CR; however, she relapsed within 6 months. She then received CAR-Ts and relapsed again, 4 months later, with diffuse adenopathy. A biopsy showed HGBCL. She received aggressive combination chemotherapy and achieved CR after 2 cycles of therapy. She was referred for consideration of allogeneic SCT (allo-SCT).
Allogeneic SCT
Allo-SCT is one of the oldest and most successful forms of cellular immunotherapies. The efficacy of allo-SCT is based, in part, on the immunologic graft-versus-lymphoma (GVL) protection provided by the donor graft.34
Allo-SCT may be curative in a subset of patients who have relapsed/refractory non-Hodgkin lymphoma (NHL) that is responsive to therapy; however, this potential cure is at the expense of high treatment-related mortality. In DLBCL, 503 patients from the Center for International Blood and Marrow Transplant Research database who relapsed after autologous SCT underwent allo-SCT and had a 5-year PFS of 29%, nonrelapse mortality (NRM) of 31%, and OS of 34%.35 Another retrospective study of 396 allo-SCT recipients with DLBCL reported that myeloablative conditioning (MAC) yielded the lowest rate of relapse at 5 years (26% vs 40% for the less intense, non-MAC treatment). In contrast, MAC had the highest rate of NRM (56% vs 36% for non-MAC).36 These results illustrate the efficacy of allo-SCT and highlight the importance of selecting an appropriate conditioning regimen based on the clinical features of individual patients.
Clinical case 2 (continued)
In our practice, allo-SCT is considered for aggressive B-cell lymphoma in patients who have relapsed/refractory disease, largely after autologous SCT and recently after CAR-T, but who have responsive disease. This patient underwent reduced-intensity, matched-sibling allo-SCT. A reduced-intensity regimen was chosen because of the extent of prior therapy, including autologous SCT. After allo-SCT, she developed grade 2, skin-only graft-versus-host disease (GVHD). The disease was successfully treated with systemic corticosteroids. All immunosuppression was tapered off within 9 months of transplantation. She had no chronic GVHD and remained disease-free 3 years after allo-SCT.
Efficacy of allogeneic SCT in other lymphomas
Allo-SCT may also be an effective cell therapy for select patients with relapsed indolent lymphoma in whom autologous SCT has failed, in whom bone marrow involvement is extensive, or in whom adequate autologous stem cells are not available.37 Compared with patients who underwent autologous SCT, patients who received allo-SCT had longer PFS (48% vs 57%, respectively), but 5-year OS was similar (72% autologous SCT vs 67% allo-SCT). These and other FL data suggest that allo-SCT is a potentially curative treatment in select patients.37 Allo-SCT has also been an effective cell therapy for select patients with other subtypes of lymphoma, including MCL37,38 and TCL.39,40
The results of donor lymphocyte infusions (DLIs) in patients who relapse after allo-SCT also provide direct evidence of GVL induction by cellular immunotherapy. In patients with relapsed lymphoma after allo-SCT, DLI is effective in a minority, although it is also associated with significant GVHD.41 More targeted cell therapies may provide antitumor activity with less nonspecific toxicity.
Future directions
Limitations and new targets
Despite the unprecedented results of autologous CAR-T, the therapy requires that each product be manufactured specifically for an individual, making manufacturing complex and expensive. The manufacturing of cells can take between 2 and 5 weeks, and may be unsuccessful. Patients with highly refractory lymphoma may develop progressive disease while awaiting CAR-T. Furthermore, heavily pretreated patients may have dysfunctional T cells, which may lead to manufacture of autologous CAR-Ts with poor efficacy.42
Moreover, at least half of patients ultimately do not have long-term responses to anti-CD19 CAR-T. Dual antigen–directed therapies are being studied to address the loss of the CD19 target antigen and to improve tumor cytotoxicity. There are multiple ways to achieve this goal, including tandem CARs (a single CAR with 2 linked single-chain variable fragments with different affinities in which T-cell activation occurs only when target cells coexpress both targets, such as CD19 and CD20), bicistronic CARs (one vector genetically modifies the T-cell to express 2 CARs, such as CD19 and CD20), and CAR pools (infusion of separately manufactured CAR-Ts with different specificities). There are also multicenter trials investigating CAR-T in combination with other active agents to increase CAR-T efficacy, such as CAR-T with checkpoint blockade, BTK inhibitors, or immunomodulatory agents (Table 3).
Selected CAR T-cell combination trials
| Clinicaltrials.gov registration no. | Trial | Lymphoma subtypes | Regimen |
|---|---|---|---|
| NCT02926833 | ZUMA-6 | DLBCL | axi-cel+atezolizumab |
| NCT03630159 | PORTIA | DLBCL | tisa-cel+pembrolizumab |
| NCT03310619 | PLATFORM | DLBCL, FL 3B, EBV+DLBCL, PMBL, HGBCL, T-cell histocyte-rich large B-cell lymphoma | liso-cel+durvalumab; liso-cel+CC-122; liso-cel+CC-220; liso-cel+ibrutinib |
| NCT03876028 | — | DLBCL | tisa-cel+ibrutinib |
| Clinicaltrials.gov registration no. | Trial | Lymphoma subtypes | Regimen |
|---|---|---|---|
| NCT02926833 | ZUMA-6 | DLBCL | axi-cel+atezolizumab |
| NCT03630159 | PORTIA | DLBCL | tisa-cel+pembrolizumab |
| NCT03310619 | PLATFORM | DLBCL, FL 3B, EBV+DLBCL, PMBL, HGBCL, T-cell histocyte-rich large B-cell lymphoma | liso-cel+durvalumab; liso-cel+CC-122; liso-cel+CC-220; liso-cel+ibrutinib |
| NCT03876028 | — | DLBCL | tisa-cel+ibrutinib |
Allogeneic CAR T cells
For patients with relapse after allogeneic SCT, donor-derived CAR-T has been effective, with minimal GVHD.43
Another allogeneic approach to CAR-T is to use a premanufactured, “off-the-shelf,” or universal, immediately available product. Donor T-cells presumably are free of prior exposure to cytotoxic therapies and may be more “fit” than a patient’s autologous T-cells. Because the cells can be premanufactured in bulk, there are also potential economic advantages.44
Theoretical complications from the use of allogeneic CAR-Ts are rapid rejection related to limited HLA matching and induction of GVHD. Gene engineering techniques are being developed and studied to produce allogeneic T cells that express a CAR while knocking out HLA expression, to limit rejection, and the native T-cell receptor, to limit the risk of GVHD.45
CAR NK cells
Allogeneic natural killer (NK) cells have been safely administered as adoptive immunotherapy for various malignancies.37 Cell engineering techniques are now available to generate and expand CAR-modified NK cells for clinical use. NK cells derived from umbilical cord blood have been engineered to express an anti-CD19 CAR with IL-15 to enhance in vivo expansion and persistence of the CAR NK cells.46 Importantly, in that study, no patient developed CRS, neurotoxicity, or GVHD. Most patients received additional therapy; thus, it is not yet possible to assess the DORs after CAR NK cells.
Conclusion
Scientific breakthroughs and rigorous clinical trials have led to rapid progress in cell therapy for B-cell lymphomas. The powerful GVL effect of allo-SCT and DLI provided evidence years ago that cellular immunotherapy could be effective for lymphoma. CAR-modified immune cells represent a more targeted, specific, and safer approach to use the immune system to treat, and likely cure, otherwise incurable lymphomas. We anticipate that CAR-T will be approved for other types of lymphoma in the future. Challenges are to decrease toxicity while improving efficacy, cost, and availability of CAR-T products. Novel transplantation approaches, allogeneic CTLs, new CAR constructs, novel target antigens, universal CAR-T or CAR NK cells, and CAR-T in combination with other agents all will be developed and tested to further harness the potential of immune cells for use in treatment of patients with lymphoma.
Acknowledgment
E.A.C. is supported by a Lymphoma Research Foundation Postdoctoral Fellowship.
