

Abstract
The overlap in clinical presentation and bone marrow features of acquired and inherited causes of hypocellular marrow failure poses a significant diagnostic challenge in real case scenarios, particularly in nonsevere disease. The distinction between acquired aplastic anemia (aAA), hypocellular myelodysplastic syndrome (MDS), and inherited bone marrow failure syndromes presenting with marrow hypocellularity is critical to inform appropriate care. Here, we review the workup of hypocellular marrow failure in adolescents through adults. Given the limitations of relying on clinical stigmata or family history to identify patients with inherited etiologies, we outline a diagnostic approach incorporating comprehensive genetic testing in patients with hypocellular marrow failure that does not require immediate therapy and thus allows time to complete the evaluation. We also review the clinical utility of marrow array to detect acquired 6p copy number-neutral loss of heterozygosity to support a diagnosis of aAA, the complexities of telomere length testing in patients with aAA, short telomere syndromes, and other inherited bone marrow failure syndromes, as well as the limitations of somatic mutation testing for mutations in myeloid malignancy genes for discriminating between the various diagnostic possibilities.
Learning Objectives
Frame a comprehensive clinical and laboratory evaluation of hypocellular marrow failure based on disease severity
Recognize that acquired 6p CN-LOH in the context of hypocellular marrow failure favors a diagnosis of aAA
Recognize that germline mutations in SAMD9, SAMD9L, and GATA2 are enriched among young patients with MDS with chromosome 7 abnormalities
Recognize the clinical utility and limitations of telomere length testing by flow-FISH in discriminating between causes of hypocellular marrow failure
Clinical case
A 28-year-old woman seeks treatment for fatigue and easy bruising. Complete blood count reveals a hemoglobin of10 g/dL, absolute neutrophil count of 1.5 × 109/L, plateletcount of 45 × 109/L, and an absolute reticulocyte count of 70 000/µL. Marrow aspirate is hypocellular for age with no frank morphologic dysplasia. Trephine biopsy specimen shows an estimated marrow cellularity of 30% and no reticulin fibrosis. Flow cytometry shows normal myeloid maturation and no abnormal or expanded myeloid blast population.
Defining hypocellular marrow failure, the differential diagnosis, and rationale for workup
Hypocellular marrow failure is characterized by peripheral cytopenia(s) and bone marrow cellularity deemed low for age and results from acquired and inherited causes that can overlap in clinical presentation and bone marrow features. For the purpose of this manuscript and to avoid ambiguity, we restrict the term aplastic anemia to the immunologically mediated disease entity, acquired aplastic anemia (aAA). Our workup centers on classifying patients as having hypocellular marrow failure requiring urgent therapy, including disease defined as severe by modified Camitta's criteria1 (Table 1) or nonsevere hypocellular marrow failure, which we define as cytopenia(s) and a hypocellular marrow for age and not meeting criteria for severe disease and who do not require urgent therapy.
Severe hypocellular marrow failure classification criteria
| Marrow cellularity |
|---|
| <25% or 25%-50% with <30% residual hematopoietic cells |
| AND cytopenias (at least 2 of 3) |
| Absolute neutrophil count <500 × 109/L Platelets <20 × 109/L Absolute reticulocyte count <60 × 109/L |
| Marrow cellularity |
|---|
| <25% or 25%-50% with <30% residual hematopoietic cells |
| AND cytopenias (at least 2 of 3) |
| Absolute neutrophil count <500 × 109/L Platelets <20 × 109/L Absolute reticulocyte count <60 × 109/L |
The immunologically mediated disease, severe aAA, accounts for the majority of severe hypocellular marrow failure.
Causes of hypocellular marrow failure are shown in Table 2. aAA is bimodal in its age at presentation, is most commonly idiopathic, and accounts for most patients with severe disease.3 Rarely and enriched among pediatric presentations, aAA is due to an underlying inborn error in immunity such as X-linked lymphoproliferative disorder.4 Accurate diagnosis of patients with nonsevere hypocellular marrow failure is clinically more challenging and requires consideration of hypocellular myelodysplastic syndrome (MDS) with or without an underlying classical inherited bone marrow failure syndrome or inherited myeloid malignancy predisposition syndrome (IBMFS/MMP). Among the most common classical IBMFSs, Fanconi anemia (FA), short telomere syndromes, and Shwachman-Diamond syndrome (SDS) may present with hypocellular marrow failure. While rare and in contrast to MDS in the elderly, childhood MDS is most commonly hypocellular,5 and also, approximately 15% of adults with MDS have a hypocellular marrow.6 Further complicating the diagnosis, patients with IBMFS/MMP (eg, mutations in GATA27 ) may have cytopenias and megakaryocytic atypia without overt malignant clonal disease that can be mistaken for MDS. Important diagnostic possibilities to consider in young persons with hypocellular MDS characterized by chromosome 7 abnormalities are GATA2 deficiency8,9 and SAMD9/9L disorders.10 While IBMFS/MMP may be suspected based on pertinent history and physical exam findings (Table 3), cryptic presentations of these disorders are increasingly recognized and thus warrant consideration even in the absence of these clues.3 Last, in all patients, the potential of a reversible cause—namely, medication-induced hypocellular marrow failure—should be excluded.
Differential diagnosis of hypocellular marrow failure
| Acquired | Inherited |
|---|---|
| aAA | Classical IBMFS/MMP Fanconi anemia Short telomere syndromes Shwachman-Diamond Syndrome GATA2 deficiency SAMD9/SAMD9L disorders |
| Anorexia nervosa | |
| Hypocellular myelodysplastic syndrome | Inborn errors of immunity (eg, X-linked lymphoproliferative disorder) |
| Medications/toxins |
| Acquired | Inherited |
|---|---|
| aAA | Classical IBMFS/MMP Fanconi anemia Short telomere syndromes Shwachman-Diamond Syndrome GATA2 deficiency SAMD9/SAMD9L disorders |
| Anorexia nervosa | |
| Hypocellular myelodysplastic syndrome | Inborn errors of immunity (eg, X-linked lymphoproliferative disorder) |
| Medications/toxins |
This table is not exhaustive and is intended to capture the more commonly encounter entities that can present as aplastic anemia. In our clinical experience, the IBMFSs/MMPs listed here are not always characterized by hypocellular marrows. Gelatinous degradation of the bone marrow also may be seen in anorexia nervosa.2
Suggested initial laboratory evaluation of hypocellular marrow failure
| Study | Source | Diagnosis | Implications for treatment |
|---|---|---|---|
| Peripheral blood | |||
| Flow cytometry for PNH | PB | aAA | IST |
| Chromosomal breakage testing | PB | FA | HSCT Modified conditioning |
| Telomere lengths by flow-FISH | PB | STS | HSCT Modified conditioning |
| Immunoglobulins, lymphocyte subsets, natural killer cell function | PB | Inborn error of immunity, GATA2 deficiency syndrome, XLP | HSCT Modified conditioning Infection prophylaxis |
| Pancreatic isoamylase | PB | SDS | |
| HLA typing on patient and full siblings | PB | Severe aAA or MDS | HSCT Donor testing |
| Bone marrow | |||
| Morphologic review of marrow | BM | MDS, GATA2 deficiency—megakaryocytic atypia | |
| Routine karyotype | BM | MDS, IBMFS/MMP | HSCT |
| FISH including −7/del, −5/del, +8, del(q20) | BM | MDS, IBMFS/MMP | HSCT |
| Chromosome genomic array | BM | 6p CN-LOH in aAA; germline CNAs in IBMFS/MMP; risk stratification of NK MDS, 7q LOH in SAMD9/9L disorders | |
| Somatic multigene genetic testing for mutations in myeloid malignancy genes | BM | See text for complexities | |
| Cultured skin fibroblasts | |||
| Germline multigene genetic testing | Fibroblasts* | IBMFS/MMP | HSCT Modified conditioning Donor testing |
| Study | Source | Diagnosis | Implications for treatment |
|---|---|---|---|
| Peripheral blood | |||
| Flow cytometry for PNH | PB | aAA | IST |
| Chromosomal breakage testing | PB | FA | HSCT Modified conditioning |
| Telomere lengths by flow-FISH | PB | STS | HSCT Modified conditioning |
| Immunoglobulins, lymphocyte subsets, natural killer cell function | PB | Inborn error of immunity, GATA2 deficiency syndrome, XLP | HSCT Modified conditioning Infection prophylaxis |
| Pancreatic isoamylase | PB | SDS | |
| HLA typing on patient and full siblings | PB | Severe aAA or MDS | HSCT Donor testing |
| Bone marrow | |||
| Morphologic review of marrow | BM | MDS, GATA2 deficiency—megakaryocytic atypia | |
| Routine karyotype | BM | MDS, IBMFS/MMP | HSCT |
| FISH including −7/del, −5/del, +8, del(q20) | BM | MDS, IBMFS/MMP | HSCT |
| Chromosome genomic array | BM | 6p CN-LOH in aAA; germline CNAs in IBMFS/MMP; risk stratification of NK MDS, 7q LOH in SAMD9/9L disorders | |
| Somatic multigene genetic testing for mutations in myeloid malignancy genes | BM | See text for complexities | |
| Cultured skin fibroblasts | |||
| Germline multigene genetic testing | Fibroblasts* | IBMFS/MMP | HSCT Modified conditioning Donor testing |
Studies recommended in all patients are bolded.
When clinically possible, cultured skin fibroblasts are the recommended DNA source for germline testing.
BM, bone marrow aspirate; NK, normal karyotype; PB, peripheral blood; PNH, paroxysmal nocturnal hemoglobinuria.
The correct diagnosis of hypocellular marrow failure is clinically paramount. Patients with severe aAA are treated with either hematopoietic stem cell transplant (HSCT) or immunosuppressive therapy (IST) with equine antithymocyte globulin and cyclosporine with or without the addition of a thrombopoietin receptor agonist.11 Marrow failure due to IBMFS/MMP or MDS is unlikely to respond to IST, and HSCT may be indicated. Recognition of a heritable disorder has important implications to the selection of an appropriate conditioning regimen, evaluation of potential related stem cell donors, and surveillance for hematopoietic and nonhematopoietic complications, as well as counseling for family members.
Clinical and laboratory evaluation of hypocellular marrow failure
A diagnostic approach to hypocellular marrow failure is shown in Figure 1. Notably, since marrow cellularity can be patchy, it is critical that the biopsy specimen be of adequate size (aim for 2 cm) and quality to ensure it is representative. Numerous guidelines detailing the recommended and suggested initial laboratory studies of hypocellular marrow failure exist.12-14 Our recommended and suggested laboratory studies are listed in Table 3. A tailored history and physical examination can be helpful in identifying the underlying cause (Table 4), particularly documentation of prior complete blood counts as longstanding cytopenias or macrocytosis can indicate a heritable cause. As IBMFS/MMP can occur without clinical stigmata or family history, an unremarkable history and examination do not reliably exclude these disorders; the presence of select disease features should raise clinical suspicion (Table 5). If an MDS-defining cytogenetic abnormality is absent, the distinction between aAA and hypocellular MDS is based on morphologic features (dysplasia and increased blasts).15 Notably, cytogenetic abnormalities can occurin aAA. Trisomy 8 and del(13q) are seen in aAA, where both carry a favorable prognosis and are associated with response to IST.16,17 Hypocellular MDS, particularly among young patients, should prompt consideration of an underlying IBMFS/MMP.
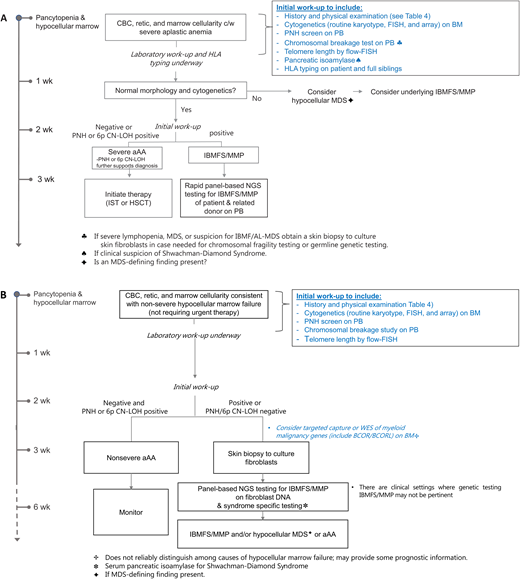
Diagnostic approach to hypocellular marrow failure based on severity. (A) Diagnostic approach to severe hypocellular marrow failure requiring therapy—time-limited. (B) Diagnostic approach to nonsevere hypocellular marrow failure not requiring therapy—not time-limited. Time-limited indicates the urgency to get the workup completed before starting therapy. BM, bone marrow aspirate; PB, peripheral blood; PNH, paroxysmal nocturnal hemoglobinuria.
Diagnostic approach to hypocellular marrow failure based on severity. (A) Diagnostic approach to severe hypocellular marrow failure requiring therapy—time-limited. (B) Diagnostic approach to nonsevere hypocellular marrow failure not requiring therapy—not time-limited. Time-limited indicates the urgency to get the workup completed before starting therapy. BM, bone marrow aspirate; PB, peripheral blood; PNH, paroxysmal nocturnal hemoglobinuria.
Focused medical history and physical examination
| Questions | Supportive information | Implications | |
|---|---|---|---|
| Presentation | Duration of symptoms | Prior CBC/diff | Inherited vs acquired |
| Bleeding | Platelet count, coagulation studies | Transfusion requirements | |
| Fatigue | Hemoglobin, pulse oximetry, CXR | Transfusion requirements | |
| S/Sx of infection | Fever, respiratory symptoms | Identify and treat infections | |
| Medical history | Adrenal hypoplasia | SAMD9 | |
| Ataxia | Brain MRI | SAMD9L | |
| Atypical infections | Immunologic testing, vaccination history | SDS, GATA2, STS, SAMD9/9L | |
| Birth history | Birth weight, gestational age, IUGR, anemia | DBA, FA, SAMD9/9L, SDS, STS | |
| Cancer or leukemia | Type of chemotherapy and toxicities | IBMFS, secondary MDS | |
| Congenital malformations | Echo, abdominal US, brain MRI, x-rays | FA | |
| GI (diarrhea, difficulty swallowing) | LFTs, abdominal US, endoscopy | aAA, SDS, STS, PNH | |
| Growth | Growth chart | FA | |
| Hearing loss | Audiogram | FA, GATA2 | |
| Nutrition (vegan, weight loss, supplements) | B12/MMA/HC, ceruloplasmin/copper | B12, folate, copper deficiency | |
| Pulmonary disease (TE fistula, fibrosis, pneumonia, alveolar proteinosis) | CXR, CT, PFTs, bronchoscopy | FA, GATA2, STS, SAMD9/9L | |
| Renal disease (congenital anomaly, dark urine) | Urinalysis, renal US | FA, PNH | |
| Skeletal dysplasia, skeletal abnormalities | X-rays | SDS, FA, DBA | |
| Family history | Ancestry | Founder mutations in certain populations | |
| Consanguinity | Autosomal recessive disorders | ||
| Full siblings | Potential marrow donors | ||
| Marrow failure, MDS, leukemia, cancer | IBMFS | ||
| Examination findings | Syndromic facies | IBMFS | |
| Microcephaly | STS, SAMD9/9L | ||
| Leukoplakia | STS | ||
| Hepatomegaly | SDS | ||
| Abnormal thumbs, radial abnormalities | FA | ||
| Nail dysplasia | STS | ||
| Lymphedema | GATA2 | ||
| Café-au-lait spots | FA | ||
| Reticulated pigmentation on neck | STS | ||
| Warts | GATA2 |
| Questions | Supportive information | Implications | |
|---|---|---|---|
| Presentation | Duration of symptoms | Prior CBC/diff | Inherited vs acquired |
| Bleeding | Platelet count, coagulation studies | Transfusion requirements | |
| Fatigue | Hemoglobin, pulse oximetry, CXR | Transfusion requirements | |
| S/Sx of infection | Fever, respiratory symptoms | Identify and treat infections | |
| Medical history | Adrenal hypoplasia | SAMD9 | |
| Ataxia | Brain MRI | SAMD9L | |
| Atypical infections | Immunologic testing, vaccination history | SDS, GATA2, STS, SAMD9/9L | |
| Birth history | Birth weight, gestational age, IUGR, anemia | DBA, FA, SAMD9/9L, SDS, STS | |
| Cancer or leukemia | Type of chemotherapy and toxicities | IBMFS, secondary MDS | |
| Congenital malformations | Echo, abdominal US, brain MRI, x-rays | FA | |
| GI (diarrhea, difficulty swallowing) | LFTs, abdominal US, endoscopy | aAA, SDS, STS, PNH | |
| Growth | Growth chart | FA | |
| Hearing loss | Audiogram | FA, GATA2 | |
| Nutrition (vegan, weight loss, supplements) | B12/MMA/HC, ceruloplasmin/copper | B12, folate, copper deficiency | |
| Pulmonary disease (TE fistula, fibrosis, pneumonia, alveolar proteinosis) | CXR, CT, PFTs, bronchoscopy | FA, GATA2, STS, SAMD9/9L | |
| Renal disease (congenital anomaly, dark urine) | Urinalysis, renal US | FA, PNH | |
| Skeletal dysplasia, skeletal abnormalities | X-rays | SDS, FA, DBA | |
| Family history | Ancestry | Founder mutations in certain populations | |
| Consanguinity | Autosomal recessive disorders | ||
| Full siblings | Potential marrow donors | ||
| Marrow failure, MDS, leukemia, cancer | IBMFS | ||
| Examination findings | Syndromic facies | IBMFS | |
| Microcephaly | STS, SAMD9/9L | ||
| Leukoplakia | STS | ||
| Hepatomegaly | SDS | ||
| Abnormal thumbs, radial abnormalities | FA | ||
| Nail dysplasia | STS | ||
| Lymphedema | GATA2 | ||
| Café-au-lait spots | FA | ||
| Reticulated pigmentation on neck | STS | ||
| Warts | GATA2 |
CBC/diff, complete blood count with differential; CT, computerized tomography; CXR, chest x-ray; GI, gastrointestinal; IUGR, intrauterine growth retardation; GATA2, GATA2 deficiency; HC, homocysteine; LFT, liver function testing; MMA, methylmalonic acid; magnetic resonance imaging; PFT, pulmonary function test; S/Sx, signs/symptoms; SAMD9/9L, SAMD9/SAMD9L disorders; TE, tracheoesophageal fistula; US, ultrasound.
Features of select inherited bone marrow failure and inherited MDS/leukemia predisposition syndromes that may present as aplastic anemia
| Syndrome | Congenital findings | Malignancy risk | Screening test | Genetics | Other hints |
|---|---|---|---|---|---|
| Fanconi anemia | Ear abnormalities, heart defects, short stature, skin pigmentation (café-au-lait spots or hypopigmentation), skeletal anomalies (thumbs, arms), TE fistula, triangular facies, urogenital defects | AML, MDS, GYN CA, head and neck CA, and others | Increased chromosome breakage | FANCA, C, G account for 95% of cases | Sensitivity to chemotherapy |
| GATA2 deficiency | Lymphedema, immunodeficiency with atypical mycobacterial infections | AML, MDS | GATA2 | Megakaryocyte atypia, pediatric MDS with monosomy 7, del7q, trisomy 8, der(1;7) | |
| SAMD9/SAMD9L disorders | MIRAGE (SAMD9): MDS, infection, restriction of growth, adrenal hypoplasia, genital phenotypes, and enteropathy Ataxia-pancytopenia syndrome (SAMD9L): cerebellar atrophy and white matter hyperintensities, gait disturbance, nystagmus | AML, MDS | SAMD9, SAMD9L | Pediatric MDS with monosomy 7, del7q, or CN-LOH 7q | |
| Short telomere syndromes | Young adults: infections, nail dystrophy, oral leukoplakia, skin hyperpigmentation Adults: emphysema, early hair graying, immune deficiency, liver fibrosis/cirrhosis, macrocytosis, pulmonary AVMs and HPS | AML, MDS Rectal adenocarcinoma, SCC anus/oral cavity/tongue | Short telomere lengths (correlates with phenotype) | DKC1, RTEL1, TERT, TERC, TINF2, RTEL1 account for majority of cases | |
| Shwachman-Diamond Syndrome | Pancreatic insufficiency (can improve with age), skeletal abnormalities | AML, MDS | Low pancreatic isoamylase (>3 years old) and low fecal elastase (pediatric and adult patients) | SBDS, SRP54, ELF1, DNAJC21 | Isolated neutropenia, somatic mutations in EIF6 and TP53 |
| Syndrome | Congenital findings | Malignancy risk | Screening test | Genetics | Other hints |
|---|---|---|---|---|---|
| Fanconi anemia | Ear abnormalities, heart defects, short stature, skin pigmentation (café-au-lait spots or hypopigmentation), skeletal anomalies (thumbs, arms), TE fistula, triangular facies, urogenital defects | AML, MDS, GYN CA, head and neck CA, and others | Increased chromosome breakage | FANCA, C, G account for 95% of cases | Sensitivity to chemotherapy |
| GATA2 deficiency | Lymphedema, immunodeficiency with atypical mycobacterial infections | AML, MDS | GATA2 | Megakaryocyte atypia, pediatric MDS with monosomy 7, del7q, trisomy 8, der(1;7) | |
| SAMD9/SAMD9L disorders | MIRAGE (SAMD9): MDS, infection, restriction of growth, adrenal hypoplasia, genital phenotypes, and enteropathy Ataxia-pancytopenia syndrome (SAMD9L): cerebellar atrophy and white matter hyperintensities, gait disturbance, nystagmus | AML, MDS | SAMD9, SAMD9L | Pediatric MDS with monosomy 7, del7q, or CN-LOH 7q | |
| Short telomere syndromes | Young adults: infections, nail dystrophy, oral leukoplakia, skin hyperpigmentation Adults: emphysema, early hair graying, immune deficiency, liver fibrosis/cirrhosis, macrocytosis, pulmonary AVMs and HPS | AML, MDS Rectal adenocarcinoma, SCC anus/oral cavity/tongue | Short telomere lengths (correlates with phenotype) | DKC1, RTEL1, TERT, TERC, TINF2, RTEL1 account for majority of cases | |
| Shwachman-Diamond Syndrome | Pancreatic insufficiency (can improve with age), skeletal abnormalities | AML, MDS | Low pancreatic isoamylase (>3 years old) and low fecal elastase (pediatric and adult patients) | SBDS, SRP54, ELF1, DNAJC21 | Isolated neutropenia, somatic mutations in EIF6 and TP53 |
AML, acute myeloid leukemia; AVM, arteriovenous malformations; CA, cancer; GYN CA, gynecological cancers; HPS, hepatopulmonary syndrome; SCC, squamous cell carcinoma; TE, tracheoesophageal fistula.
Diagnostic testing in hypocellular marrow failure requiring therapy (Figure 1A) is done simultaneously, rather than sequentially, to allow definitive treatment to start expeditiously to limit transfusions and the risk of serious infection. We aim to initiate treatment within approximately 3 weeks of presentation, which allows time to complete diagnostic studies and donor evaluations. Evaluation of hypocellular marrow failure not requiring urgent therapy can typically proceed in a more stepwise fashion (Figure 1B). There are clinical settings where genetic testing for IBMFS/MMP may not be pertinent (eg, elderly patient with multiple comorbidities and no potentially affected family members in whom testing would not inform the patient's or family's care).
CLINICAL CASE (continued)
Old records are requested and document several normal past complete blood counts. Medical history and examination are unremarkable. Chromosomal breakage studies return unremarkable. Telomere length testing falls in the 30th age-adjusted percentile.
Chromosomal breakage test
Given the impact a diagnosis of FA has on treatment and surveillance strategies, a chromosomal breakage test is indicated in all patients with hypocellular marrow failure. Abnormal test results should be followed with genetic testing of a germline tissue source to try to identify the causative mutation as this provides additional prognostic information regarding cancer and other risks as well as facilitates carrier testing for family members.18 Breakage studies allow diagnosis of patients whose mutations are missed by standard genetic testing and inform the functional consequence of genetic variants that might otherwise be of unclear clinical significance. This testing is performed by culturing either peripheral blood lymphocytes or skin fibroblasts in the presence of DNA cross-linking agents and comparing the number of chromosomal breaks, including rearrangements, gaps, endoreduplications, and exchanges, to controls under baseline and stimulated conditions. Results are reported as both the number of cells with abnormal breaks and the number of breaks per cell. Although peripheral blood lymphocytes are the most accessible source of tissue for breakage and genetic studies, lymphocytes are susceptible to the phenomenon of somatic mosaicism resulting from expansion of hematopoietic stem cells that have undergone molecular reversion that corrects the FA phenotype. This is estimated to occur in approximately 10% to 25% of patients with FA.19 Somatic reversion may be suspected if fragility testing performed on lymphocytes shows 2 populations of cells, some appearing normal and others with multiple breaks per cell. Negative results should be confirmed in cultured fibroblasts if FA is strongly suspected. Breakage studies on fibroblasts can also be considered when studies in lymphocytes fail due to severe leukopenia or poor growth in culture.
Complexities of telomere length testing
The short telomere syndromes are a group of genetic disorders that are caused by mutations in components of the telomerase enzyme and other telomere maintenance genes. Telomere length testing by flow cytometry and fluorescence in situ hybridization (flow-FISH) is the gold standard because of its high reproducibility. It measures single-cell telomere length using a fluorescently labeled probe that hybridizes to telomere DNA. Clinical testing is performed on a peripheral blood sample, and available assays report results in total lymphocytes and granulocytes (so called 2-panel testing) or granulocytes and total lymphocytes and lymphocyte subsets (naive T cells, memory T cells, B cells, natural killer cells), so-called 6-panel testing. Two-panel testing appears sufficient for the initial diagnostic workup of a patient with hypocellular marrow failure.20
Telomere length (TL) assessment is recommended in the initial diagnostic workup of hypocellular marrow failure. TL assessment is useful in ruling out a diagnosis of a constitutional short telomere syndrome upfront in patients with marrow failure. Lymphocyte telomere length above the age-adjusted 50th percentile has a 100% negative predictive value in identifying a clinically meaningful mutation in a short telomere syndrome (STS) gene in pediatric and adult patients.21 Among patients younger than 40 years, a lymphocyte telomere length below the age-adjusted first percentile strongly supports a diagnosis of a STS but lack specificity.21,22 Telomere lengths below the age-adjusted first percentile have been reported in patients with other IBMFSs, including FA and SDS,21,23 and so consideration of the clinical presentation and additional genetic testing results are necessary to establish the correct diagnosis. In a predominantly pediatric and younger adult cohort of patients younger than 40 years (0-31 years; median, 14 years) presenting with idiopathic marrow failure, all of the patients with immunotherapy-responsive marrow failure had lymphocyte telomere lengths above the age-adjusted first percentile.21 Nearly half of the patients with genetically confirmed short telomere syndromes older than 40 years have telomere length measurements above the age-adjusted first percentile, and so telomere lengths above this threshold do not exclude the diagnosis of a short telomere syndrome in older patients and requires diagnosis here additional clinical and genetic information.21,22 Telomere length assessment in various causes of hypocellular marrow failure are shown in Figure 2.
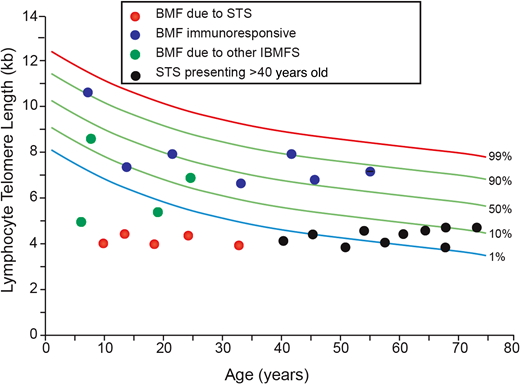
Utility of telomere length testing in the diagnosis of hypocellular marrow failure. The red circles denote patients with bone marrow failure secondary to a genetically confirmed diagnosis of a short telomere syndrome. The blue circles denote patients with presumed idiopathic acquired aplastic anemia based on response to treatment with IST. The green circles denote patients with bone marrow failure due to an underlying genetically confirmed inherited bone marrow failure syndrome apart from a short telomere syndrome (LIG4,RUNX1,GATA2). The black circles denote patients diagnosed with a short telomere syndrome at >40 years of age. BMF, bone marrow failure. Based on data from Alder et al.21
Utility of telomere length testing in the diagnosis of hypocellular marrow failure. The red circles denote patients with bone marrow failure secondary to a genetically confirmed diagnosis of a short telomere syndrome. The blue circles denote patients with presumed idiopathic acquired aplastic anemia based on response to treatment with IST. The green circles denote patients with bone marrow failure due to an underlying genetically confirmed inherited bone marrow failure syndrome apart from a short telomere syndrome (LIG4,RUNX1,GATA2). The black circles denote patients diagnosed with a short telomere syndrome at >40 years of age. BMF, bone marrow failure. Based on data from Alder et al.21
CLINICAL CASE (continued)
Marrow karyotype returns 46,XX. Microarray on the marrow aspirate sample identified copy number-neutral loss of heterozygosity of 6p in 10% of the cells. Flow cytometry on peripheral blood did not identify any glycosylphosphatidylinositol (GPI)-deficient populations.
Diagnostic utility of select genetic abnormalities in hypocellular marrow failure
Acquired copy number-neutral loss of heterozygosity of chromosome arm 6p and chromosomal microarray testing on platforms including single-nucleotide polymorphism probes
More than 70% of patients (and over 60% of pediatric patients) with aAA have clonal hematopoiesis with somatic mutations detected by whole-exome sequencing or single-nucleotide polymorphism array early in the course of their disease.24,25 Clonal hematopoiesis does not equate to malignancy and reflects an acquired genetic change in a hematopoietic stem cell, allowing for competitive outgrowth of the mutated hematopoietic stem cell's progeny. The 2 most common somatic mutations in aAA are mutational inactivation of PIGA and the loss of HLA class I alleles. The clinical utility of detecting PIGA loss or the functional consequence of a detectable paroxysmal nocturnal hemoglobinuria clone in hypocellular marrow failure is covered in Education Session entitled “When does a PNH clone have clinical significance?”26 PIGA loss is most commonly diagnosed by flow-cytometric analysis to detect cells lacking GPI-linked proteins. This study is more sensitive when performed on a peripheral blood sample compared with a bone marrow aspirate sample as the GPI-linked proteins typically assessed are most highly expressed on peripheral blood cells. Thus, testing on peripheral blood as opposed to a marrow sample for a paroxysmal nocturnal hemoglobinuria clone is recommended.27
Loss of HLA class I alleles occurs either through a loss-of-function mutation in 1 of the HLA class I genes or, more commonly, through the loss of 1 parental HLA haplotype through the acquisition of copy number-neutral loss of heterozygosity of chromosome arm 6p (6p CN-LOH) involving the human leukocyte antigen locus. A prevailing hypothesis is that the loss of HLA class I alleles disrupts antigen presentation, allowing nondominant clones to evade the immune system. An analogous pathophysiology may contribute to posttransplant relapse in haploidentical HSCT recipients, in whom the relapsed disease has acquired loss of the HLA haplotype that differed from the donor's haplotype, thus dampening the graft-versus-leukemia effect.26,28,29 In aAA, the HLA alleles specifically lost through 6p CN-LOH vary across age and ethnic origin of patients. That said, studies report a bias of the missing HLA allele in 6p CN-LOH to particular HLA types. This, together with the finding that the HLA allele is commonly involved across the 6p CN-LOH reported in aAA, suggests the finding is linked to the pathogenesis of aAA and not merely a secondary event. Although additional studies are needed to clarify the impact of acquired 6p CN-LOH on response to IST, the presence of 6p CN-LOH appears to be diagnostically informative in distinguishing patients with immune-mediated marrow failure from those with inherited bone marrow failure conditions.30-33 Acquired 6p CN-LOH appears to be relatively specific for aAA, occurring in less than 1% of patients with MDS or in normal aging.30,34,35 The prevalence of clonal 6p CN-LOH among pediatric and adult patients with aAA is approximately 10% to 13%.24,33,36 Constitutional 6p homozygosity is not enriched among patients with aAA.
Clinical testing for 6p CN-LOH requires chromosomal microarray analysis (CMA). Outside of detecting 6p CN-LOH, additional rationale supporting inclusion of CMA in the workup of hypocellular marrow failure includes that this testing can be performed on DNA extracted from residual cell pellets, aspirate smears, touch preps, or formalin-fixed, paraffin embedded tissues. Cells obtained from patients with hypocellular marrow failure may not grow well in culture for karyotypic analysis. In addition to detecting CN-LOH, CMA can identify microdeletions and microduplications beyond the resolution of conventional karyotype and FISH,37 potentially providing a rationale for closer clinical surveillance for malignant clonal evolution. In addition, CMA may detect copy number alterations (CNAs) missed by some targeted next-generation sequencing (NGS)–based testing. Causative CNAs have been reported in a number of IBMFs/MMPs, including FA and others.38,39 In 1 cohort study of patients with suspected inherited bone marrow failure, CMA identified germline pathogenic CNAs in 16.4% (11/67) of patients tested.40
Somatic mutation testing for mutations in myeloid malignancy genes
The profile of somatic mutations identified by NGS strategies in aAA, pediatric MDS, hypocellular MDS in adults, and IBMFS/MMP differs from one another. An understanding of this context-dependent clonal landscape and its dynamics continues to provide important insights into malignant and nonmalignant clonal evolution and, in select settings, offers prognostic insights. This has been recently reviewed.40-42 The current diagnostic utility of NGS panels and whole-exome sequencing for somatic mutations in myeloid malignancy genes to reliably distinguish among these hypocellular marrow failure syndromes in the clinic remains unclear. A specific somatic mutational landscape that can reliably identify bona fide MDS (in the absence of definitive morphologic or cytogenetic findings) for clinical practice in this setting has not been established. Approximately 25% to 30% of patients with aAA have clonal mutations in genes that are associated with both myeloid malignancy and with aging, including ASXL1, DNMT3A, JAK2, and TP53. These mutations are present at low variant allele frequency similar to what is seen in age-related clonal hematopoiesis, and these genes can also be mutated in adult hypocellular MDS.24,43,44 In addition, although mutations in the epigenetic modifier genes, BCOR and BCORL1, appear subtly enriched in aAA compared with hypocellular MDS, both diseases can carry these mutations, and so this finding is not sufficient to distinguish between these disorders.44 In contrast to typical adult MDS, mutations in the spliceosome complex (ie, SF3B1, SRSF2, ZRSR2, U2AF1) are rare across refractory cytopenia of childhood,9,11 adult hypoplastic MDS,44,45 and aAA,24,44 so this finding is also not informative diagnostically. When feasible, this testing can be considered for its potential prognostic value.46 As examples, the acquisition of biallelic TP53 mutations is associated with development of leukemia in SDS,47 and in aAA, BCOR and BCORL1 mutations are associated with response to immunosuppression and a favorable prognosis.24
CLINICAL CASE (continued)
Based on her workup, the patient was diagnosed with nonsevere aAA and is being followed by serial blood counts.
Conclusion
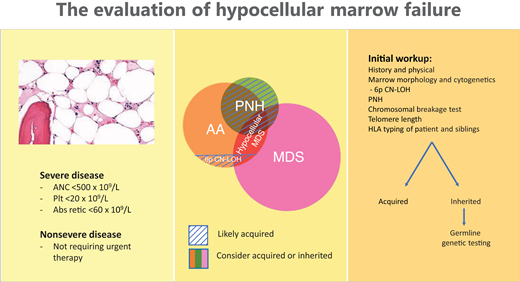
The evaluation of hypocellular marrow failure (Figure 3) is based on disease severity and aims to distinguish among the inherited and acquired causes to inform optimal patient care.
Acknowledgments
We thank patients and families for participating in marrow failure research and Mary Armanios at Johns Hopkins University for providing Figure 2.
Conflict-of-interest disclosures
Amy Geddis: no relevant conflicts of interest.
Siobán Keel: no relevant conflicts of interest.
Off-label drug use
Amy Geddis: nothing to disclose.
Siobán Keel: nothing to disclose.
