

Abstract
Paroxysmal nocturnal hemoglobinuria (PNH) is an acquired blood disease caused by somatic mutations in the phosphatidylinositol glycan class A (PIGA) gene required to produce glycophosphatidyl inositol (GPI) anchors. Although PNH cells are readily identified by flow cytometry due to their deficiency of GPI-anchored proteins, the assessment of the clinical significance of a PNH clone is more nuanced. The interpretation of results requires an understanding of PNH pathogenesis and its relationship to immune-mediated bone marrow failure. Only about one-third of patients with PNH clones have classical PNH disease with overt hemolysis, its associated symptoms, and the highly prothrombotic state characteristic of PNH. Patients with classical PNH benefit the most from complement inhibitors. In contrast, two-thirds of PNH clones occur in patients whose clinical presentation is that of bone marrow failure with few, if any, PNH-related symptoms. The clinical presentations are closely associated with PNH clone size. Although exceptions occur, bone marrow failure patients usually have smaller, subclinical PNH clones. This review addresses the common scenarios that arise in evaluating the clinical significance of PNH clones and provides practical guidelines for approaching a patient with a positive PNH result.
Learning Objectives
Interpret the validity and significance of PNH flow cytometry results
Recognize the significance of PNH clones in the diagnostic assessment of patients with bone marrow failure
Recognize indications for starting complement inhibitor therapy in PNH
Introduction
Paroxysmal nocturnal hemoglobinuria (PNH) is an acquired blood disease caused by somatic mutations in the phosphatidylinositol glycan class A (PIGA) gene.1 PIGA encodes an enzyme required for the first step of glycophosphatidyl inositol (GPI) anchor biosynthesis. PIGA-mutant (PNH) cells lack all GPI-anchored proteins, including 2 essential complement regulatory proteins, CD55 (complement decay-accelerating factor) and CD59 (inhibitor of the complement membrane attack complex). The deficiency of CD55 and CD59 makes PNH red blood cells (PNH RBCs) susceptible to complement-mediated hemolysis.
Although very small polyclonal populations of PNH cells of approximately 0.001% to 0.005% (approximately 10 to 50 per million) can be detected in most healthy individuals,2,3 PNH hematopoietic cells have no intrinsic growth advantage and do not clonally expand under normal conditions.4-6 Slightly larger PNH clones of 0.02% to 0.03% can occur in approximately 1% of healthy individuals; however, more significant clonal expansions of PNH cells do not occur in the absence of pathology.2,3,7 In contrast, PNH clones are prevalent in patients with immune-mediated bone marrow failure.8-10 Nearly half of patients with acquired aplastic anemia (AA), a T-cell–mediated autoimmune bone marrow failure disorder, develop PNH clones.8-10 In AA, PNH cells are thought to evade the hematopoietic stem and progenitor cell (HSPC)-directed autoimmune attack, and consequently, PNH cells outgrow wild-type cells forming PNH clones.1 PNH clones can also emerge in patients with myelodysplastic syndrome (MDS) and, more rarely, in patients with myeloproliferative neoplasms.11,12 Although immune-mediated bone marrow failure is the primary risk factor for developing PNH clones, patients can be diagnosed with classical PNH without a known diagnosis of bone marrow failure or a history of cytopenias. In such cases, prior subclinical marrow failure is frequently presumed; alternatively, secondary proliferative mutations within a PNH HSPC can promote PNH clone expansion.1,13
Studies of patients with positive PNH flow cytometry results have found that PNH clones have a bimodal size distribution closely related to the patients' clinical presentations12,14 (Figure 1, Table 1). Approximately one-third of patients have “classical PNH,” with overt hemolysis, multiple PNH symptoms, and an increased risk of thromboses. Patients with classical PNH have a mean PNH clone size of more than 70%.11,12 In contrast, two-thirds of PNH clones occur in patients who have cytopenias and bone marrow failure without clinical hemolysis. Most bone marrow failure patients are largely asymptomatic from PNH and have small PNH clones (mean clone size of approximately 11%) (Figure 1, Table 1).11,12
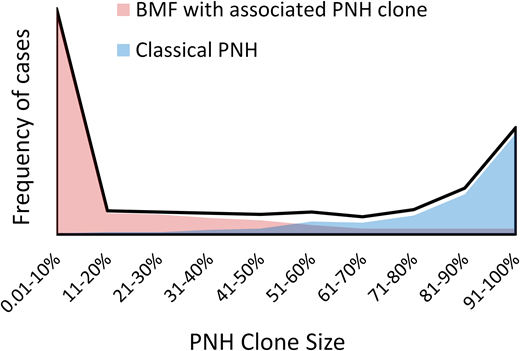
The bimodal distribution of PNH clone sizes closely correlates with the patients' clinical presentations. The stylized schematic depicts the bimodal distribution of granulocyte PNH clone sizes (black line) based on published PNH clone size distributions.12,14 Red and blue area plots illustrate the different clinical presentations that correlate with the extremes of PNH clone sizes. Patients with bone marrow failure who have an associated PNH clone are shown in red. Classical PNH presentations are shown in blue.
The bimodal distribution of PNH clone sizes closely correlates with the patients' clinical presentations. The stylized schematic depicts the bimodal distribution of granulocyte PNH clone sizes (black line) based on published PNH clone size distributions.12,14 Red and blue area plots illustrate the different clinical presentations that correlate with the extremes of PNH clone sizes. Patients with bone marrow failure who have an associated PNH clone are shown in red. Classical PNH presentations are shown in blue.
Clinical categories of patients with PNH clones
| PNH category | PNH granulocyte clone | Clinical hemolysis | Laboratory markers of hemolysis | PNH symptoms | Risk of thrombosis | Utility of complement inhibitor therapy |
|---|---|---|---|---|---|---|
| Bone marrow failure with an associated PNH clone | ||||||
| Subclinical (majority of patients) | Small (usually <30%) | Absent | May be present, depending on clone size | Absent | Low | Do not benefit from complement inhibitor |
| Symptomatic | Moderate | May be present | May be present | May be present | Intermediate | Some patients may benefit from complement inhibitor |
| Classical PNH | Large (usually >50%) | Present | Present | Present | High | Complement inhibitor improves outcomes |
| PNH category | PNH granulocyte clone | Clinical hemolysis | Laboratory markers of hemolysis | PNH symptoms | Risk of thrombosis | Utility of complement inhibitor therapy |
|---|---|---|---|---|---|---|
| Bone marrow failure with an associated PNH clone | ||||||
| Subclinical (majority of patients) | Small (usually <30%) | Absent | May be present, depending on clone size | Absent | Low | Do not benefit from complement inhibitor |
| Symptomatic | Moderate | May be present | May be present | May be present | Intermediate | Some patients may benefit from complement inhibitor |
| Classical PNH | Large (usually >50%) | Present | Present | Present | High | Complement inhibitor improves outcomes |
Interpreting the significance of a PNH flow cytometry result
CLINICAL CASE 1: FALSE-POSITIVE PNH TEST
A 55-year-old man with decades-long thrombocytopenia was referred for an evaluation of bone marrow failure. The patient had macrocytic anemia with a hemoglobin of 10.6 g/dL and a mean corpuscular volume of 121.4 fL, an absolute reticulocyte count of 45.8 × 103 cells/µL, and platelets of 29 × 103 cells/µL. The white blood cells (WBCs) were low at 2.9 × 103 cells/µL with 58.9% granulocytes, 6.2% monocytes, and 34.9% lymphocytes. Bone marrow was hypocellular without dysplastic changes. PNH flow cytometry revealed an isolated PNH clone of 2.9% in RBCs only, with no PNH granulocytes. Lactate dehydrogenase (LDH) was normal at 191 U/L. Haptoglobin was borderline low at 32 mg/dL. Given the absence of granulocyte PNH clone and the lack of hemolysis, a false-positive PNH clone was suspected. PNH flow cytometry testing was repeated, with no evidence of PNH cells on several subsequent tests. Additional workup showed short lymphocyte telomere lengths for age and a pathogenic variant in TERC, confirming the diagnosis of short telomere syndrome.
The diverse clinical presentations and varied PNH cell type involvement require systematic consideration of several clinical and laboratory factors to determine the significance of a “positive” (or a “negative”) PNH flow cytometry result for a given patient. At a minimum, an evaluation of a PNH result should include (1) PNH flow cytometry, which should be performed on peripheral blood with quantification of PNH neutrophils and PNH RBCs; (2) a bone marrow aspirate and biopsy, including cytogenetics (testing for somatic mutations associated with hematologic malignancies can also be helpful for prognostic assessment of patients with hypocellular marrow failure13 and in patients in whom PNH arose in association with MDS or myeloproliferative neoplasms); (3) laboratory studies to evaluate for hemolysis; and (4) history assessing for prior thrombotic events and cytopenias (Figure 2). Here, I discuss several common clinical scenarios that arise when evaluating patients with a positive PNH test to illustrate the significance of PNH clones in classical PNH and bone marrow failure.
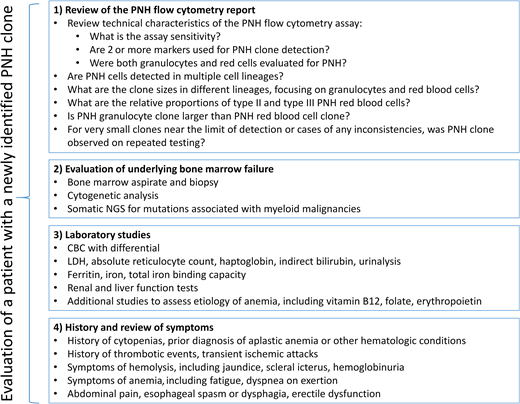
The recommended evaluation algorithm for patients with a newly identified PNH clone. CBC, complete blood count; NGS, next-generation sequencing.
The recommended evaluation algorithm for patients with a newly identified PNH clone. CBC, complete blood count; NGS, next-generation sequencing.
PIGA mutations occur early in hematopoiesis in hematopoietic stem cells or multipotent progenitors.7,12,14,15 In most patients, PNH clones can be detected in all or most mature hematopoietic lineages, including granulocytes, monocytes, platelets, RBCs, and the B, T, and natural killer cells.7,12,15 More limited lineage involvement can occur but is more characteristic of MDS, in which PNH clones are also more likely to be transient.7
Next-generation sequencing of the PIGA gene in patients with PNH identified 2 or more independent PIGA mutations in 85% of patients with PNH and at least 3 independent PIGA mutations in 57% of patients.16 Because multiple independent mutations together comprise PNH clones detected by flow cytometry, flow cytometry is much more sensitive than next-generation sequencing for PNH detection. In contrast to genetic analyses, which are more sensitive when performed on bone marrow than peripheral blood by capturing acquired genetic alterations in HSPCs, the standard clinical PNH flow cytometry evaluates only the mature cell populations (granulocytes, monocytes, and RBCs) that are well represented in peripheral blood. PNH clones can thus be sensitively detected in peripheral blood without a requirement for a bone marrow biopsy. Sometimes, submitting a hypocellular bone marrow specimen for PNH flow cytometry may paradoxically result in lower sensitivity for PNH detection if the submitted aspirate is of an inadequate volume containing insufficient granulocytes to detect very small PNH clones. While PNH flow cytometry of other cell populations in the bone marrow can be performed, these have not been standardized for high sensitivity PNH cell analysis and can be affected by variation in cell surface marker expression across various hematopoietic subsets.
Clinically, most flow cytometry laboratories now use high-sensitivity PNH assay, allowing the detection of PNH clones as small as 0.01% in granulocyte, monocyte, and RBC lineages. PNH granulocyte and monocyte proportions are closely correlated in most patients.11,12 Unlike PNH RBCs, which are susceptible to complement-mediated hemolysis, PNH granulocytes have a normal life span in vivo.17 For this reason, the PNH clone size is commonly estimated using the granulocyte lineage. At a minimum, PNH flow cytometry should provide a quantification of PNH proportions in granulocytes and RBCs.
To ensure that the identified GPI-negative cell populations represent PNH clones and are not cells with aberrant expression or an inherited deficiency of a single GPI-anchored protein,18 at least 2 GPI-anchored markers should be used to identify PNH cells. The incorporation of the fluorochrome-conjugated nonlysing form of the proaerolysin toxin, which binds to the glycan moiety of GPI-anchored proteins, has further improved the sensitivity and accuracy of PNH leukocyte detection.19 Granulocyte and monocyte PNH clones are quantified based on complete GPI-anchored protein deficiency. In contrast, both the complete (type III) and the partial (type II) GPI-anchored protein deficiency are measured for RBCs. Type II cells have a small fraction of normal GPI-anchored protein levels and arise from either hypomorphic (missense) PIGA mutations or the passive transfer of GPI-anchored proteins to PNH cells.16,20 Rarely, type II cells may significantly outnumber type III cells, in which case the underlying PIGA mutation is commonly a missense variant in PIGA, leading to a hypofunctioning allele and a partial GPI-anchored protein deficiency.16
In patients not receiving complement inhibitors, PNH RBCs are sensitive to complement-mediated lysis and have a half-life of only 4 to 8 days.21 The shortened life span of PNH RBCs causes a significantly smaller apparent RBC PNH clone size compared with PNH granulocytes. A corollary to this is that the lack of the expected clone size difference between PNH granulocytes and RBCs in an untreated patient with PNH implies a normal PNH RBC life span and a lack of clinically significant hemolysis; in practice, this can arise due to a predominant type II clone that is less susceptible to hemolysis. Because RBC transfusions will dilute the PNH RBC fraction, a transfusion history should be taken into account when interpreting PNH RBC clone size. When clinically feasible, PNH flow cytometry should be performed before RBC transfusions.
Most PNH clones persist long term, of which some (approximately 6% to 18%) will expand, while 17% to 24% of clones will become smaller over time.9,10,12,14,22 Thus, serial evaluation (eg, annually and with changes in hemolytic parameters) is recommended for all patients with PNH clones to monitor for PNH clone expansion. Periodic monitoring can also be helpful in patients with classical PNH, up to 15% of whom can undergo spontaneous remission over time and may no longer require therapy.13 Because most PNH clones arise in patients with AA, annual PNH screening is advised for patients with AA to identify newly emergent PNH clones.23
Reflecting on case 1, the pattern of an isolated PNH RBC clone without the involvement of granulocytes is unexpected. This pattern raises a possibility of a false-positive PNH clone or, perhaps, a transient single-lineage PNH clone isolated to the erythroid lineage. Clinically, the patient has no laboratory or clinical hemolysis, which is not necessarily unexpected given the relatively small RBC clone size. Because PNH cells are identified by their lack of staining for surface markers, specialized flow cytometry protocols are required to prevent falsely misidentifying debris and other nonstaining cells as PNH clones.24 Historically, failure to use optimal methodology has been associated with false-positive results in 16% to 25% of clinical PNH tests in multicenter comparisons.11,25 The implementation of the International Clinical Cytometry Society/European Society for Clinical Cell Analyses consensus guidelines for detection of GPI-deficient cells in PNH has dramatically improved the accuracy and sensitivity of PNH testing. Compared with routine PNH analysis suitable for identifying PNH clones more than 1%, which misses nearly 40% of smaller PNH clones,19 the high sensitivity PNH assay enables accurate identification of clones down to 0.01%.19,24 Because rare healthy individuals can have transient expansions of PNH cells reaching the level of detection of the high-sensitivity PNH assay,2,3,7 repeating PNH flow cytometry can help to ascertain persistence of very rare populations of PNH cells.
In sum, the technical characteristics of a given PNH flow cytometry assay can significantly affect the accuracy of results. A careful review of the PNH testing methodology, with close attention to the assay sensitivity and specificity parameters, the presence of PNH cells in multiple lineages, and the expected difference between the granulocyte and RBC PNH clone sizes, can help interpret the accuracy of both the positive and negative PNH results in a given patient (Figure 2). Follow-up testing with high-sensitivity PNH assay, performed according to the International Clinical Cytometry Society/European Society for Clinical Cell Analyses guidelines, can clarify unexpected results and resolve inconsistencies.
PNH clones in immune-mediated bone marrow failure
CLINICAL CASE 2: DIAGNOSTIC SIGNIFICANCE OF PNH CLONES IN AA
A 26-year-old previously healthy woman sought treatment from the emergency room after experiencing 1 month of fatigue and dyspnea on exertion. Complete blood count revealed pancytopenia, with WBCs of 1.07 × 103 cells/µL, with 24.3% neutrophils and 75.7% lymphocytes, a hemoglobin of 4.3 g/dL with low absolute reticulocytes of 23 × 103 cells/µL, and platelets of 12 × 103 cells/µL. Physical examination was notable for pallor and scattered petechiae. There was no family history of blood count abnormalities or conditions suspicious for inherited marrow failure syndromes. Additional laboratory studies showed a mildly elevated LDH of 239 U/L (1.2 × the upper limit of normal [ULN] in the testing laboratory), a low haptoglobin less than 30 mg/dL, and normal indirect bilirubin of 0.9 mg/dL. The marrow was markedly hypocellular for age without dysplastic changes. Cytogenetics were normal. Flow cytometry revealed a PNH clone of 19.11% in neutrophils, 17.83% in monocytes, and 0.32% in RBCs. A diagnosis of severe immune-mediated AA with an associated PNH clone was made. The patient had no matched sibling donors and was treated with immunosuppressive therapy with horse antithymocyte globulin, cyclosporine, and eltrombopag, achieving complete remission. One year after initial diagnosis, a follow-up evaluation revealed a hemoglobin of 11.9 g/dL and 48 × 103 reticulocytes/µL with no clinical or laboratory evidence of hemolysis that included a normal LDH of 186 U/L. Follow-up PNH flow cytometry showed a smaller PNH clone of 8.26% in neutrophils, 7.6% in monocytes, and 1.59% in RBCs.
Because the clonal expansion of PNH cells is closely linked to the HSPC-directed autoimmune attack in AA,1 a PNH clone can provide an important diagnostic clue about the immune-mediated pathogenesis of marrow aplasia.8,26 The presenting features of AA—pancytopenia with hypocellular bone marrow—can be brought on by different etiologies and are not specific for the diagnosis of AA. For this reason, the diagnosis of AA requires systematic exclusion of all alternative causes of marrow failure, including nutritional deficiencies, infections, or toxins. Of particular importance for children and young and middle-aged adults is the exclusion of inherited bone marrow failure syndromes, a costly and time-consuming process that can leave lingering diagnostic uncertainties. Consequently, clinical testing with a high positive predictive value for immune-mediated AA can be valuable by streamlining the diagnostic evaluation and allowing for prompt initiation of AA-directed therapies.
The predictive value of PNH clones for the diagnosis of immune-mediated AA and the exclusion of inherited bone marrow failure was evaluated in 2 institutional cohorts of bone marrow failure patients8,26 (Table 2). PNH clones were identified in 46% of patients with AA but in none of the evaluated inherited bone marrow failure patients. A combined analysis of both cohorts showed that PNH granulocyte clones, regardless of clone size, have an approximately 100% positive predictive value for AA and have 25-fold higher odds of being found in AA compared with inherited bone marrow failure.8 Two additional studies found no PNH clones or PIGA mutations in more than 130 unselected patients with an inherited marrow failure disorder, Shwachman-Diamond syndrome (SDS),27,28 providing additional support to the specificity of PNH clones for immune-mediated AA.
Frequency of PNH in AA compared with inherited bone marrow failure disorders
| Study | Year | Population | Study design | Disease: number of patients | PNH testing method | Frequency of PNH clones | Comparison |
|---|---|---|---|---|---|---|---|
| DeZern et al26 | 2014 | JHU cohort | Retrospective | AA: n = 132 IBMF: n = 20 | Flow cytometry on peripheral blood (sensitivity range >0.01%-0.1%) | AA: 61 of 132 (46%) IBMF: 0 of 20 (0%) | • PPV for AA: 100% • NPV for AA: 54% |
| Shah et al8 | 2021 | Penn/CHOP cohort | Retrospective | AA: n = 126 Inherited: n = 9 Other: n = 13 | Flow cytometry on peripheral blood (sensitivity range >0.01%-1%) | AA: 58 of 126 (46%) Inherited and other: 0 of 22 (0%) | • AA vs IBMF, OR 11.10, P < .05 • AA vs all inherited disorders including IBMF, OR 16.23, P < .05 • AA vs all non-AA, non-PNH, OR 38.43, P < .05 • PPV for AA vs IBMF: 100% • NPV for AA vs IBMF: 48.5% |
| Keller et al27 | 2002 | Camp Sunshine SDS cohort | Retrospective | n = 28 | Flow cytometry on peripheral blood (PNH clones >1%) | 0 of 28 patients (0%) | NA |
| Kennedy et al28 | 2014 | SDS cohort | Retrospective | SDS: n = 99 SDS-like: n = 11 | Targeted, error-corrected sequencing of PIGA in bone marrow (>0.1% VAF) | 0 of 110 patients with PIGA mutations (0%) | NA |
| Study | Year | Population | Study design | Disease: number of patients | PNH testing method | Frequency of PNH clones | Comparison |
|---|---|---|---|---|---|---|---|
| DeZern et al26 | 2014 | JHU cohort | Retrospective | AA: n = 132 IBMF: n = 20 | Flow cytometry on peripheral blood (sensitivity range >0.01%-0.1%) | AA: 61 of 132 (46%) IBMF: 0 of 20 (0%) | • PPV for AA: 100% • NPV for AA: 54% |
| Shah et al8 | 2021 | Penn/CHOP cohort | Retrospective | AA: n = 126 Inherited: n = 9 Other: n = 13 | Flow cytometry on peripheral blood (sensitivity range >0.01%-1%) | AA: 58 of 126 (46%) Inherited and other: 0 of 22 (0%) | • AA vs IBMF, OR 11.10, P < .05 • AA vs all inherited disorders including IBMF, OR 16.23, P < .05 • AA vs all non-AA, non-PNH, OR 38.43, P < .05 • PPV for AA vs IBMF: 100% • NPV for AA vs IBMF: 48.5% |
| Keller et al27 | 2002 | Camp Sunshine SDS cohort | Retrospective | n = 28 | Flow cytometry on peripheral blood (PNH clones >1%) | 0 of 28 patients (0%) | NA |
| Kennedy et al28 | 2014 | SDS cohort | Retrospective | SDS: n = 99 SDS-like: n = 11 | Targeted, error-corrected sequencing of PIGA in bone marrow (>0.1% VAF) | 0 of 110 patients with PIGA mutations (0%) | NA |
IBMF, inherited bone marrow failure; Inherited, inherited hematologic disorders; JHU, Johns Hopkins University; NA, not applicable; NPV, negative predictive value; OR, odds ratio; Penn/CHOP, University of Pennsylvania/Children's Hospital of Philadelphia; PPV, positive predictive value; VAF, variant allele fraction.
In clinical practice, a Bayes theory nomogram tool, which incorporates the pretest probability of AA, can be used to estimate the posttest probability of AA after a positive PNH result (Figure 3).8 In Case 2, a previously healthy patient developed pancytopenia as a young adult, without a family history or physical examination findings suggestive of a congenital marrow failure disorder. Based on this presentation, the clinical suspicion of immune-mediated acquired AA is moderate to high. Using a conservative estimate of a moderate pretest probability of AA (eg, 60%), the presence of a PNH clone would raise the posttest probability of AA to more than 99%, allowing to confidently establish the diagnosis of immune-mediated AA and initiate appropriate therapy. Notably, a negative PNH test does not significantly change the pretest probability of AA, and the absence of a PNH clone should not be used to exclude AA. PNH testing can similarly help in situations where a priori clinical suspicion of AA is low. For example, a diagnosis of AA may be discounted in a young adult with a known syndromic disorder with multiple congenital anomalies and moderate cytopenias from birth who developed worsening cytopenias and new transfusion requirements following interferon therapy for hepatitis C. Finding a PNH clone, in this or similar situations of poorly explained cytopenias, can bring AA higher on the differential diagnosis. In a hypothetical case with a low pretest probability of 2%, a positive PNH clone would raise the posttest probability of AA into a moderate range of 65%.8
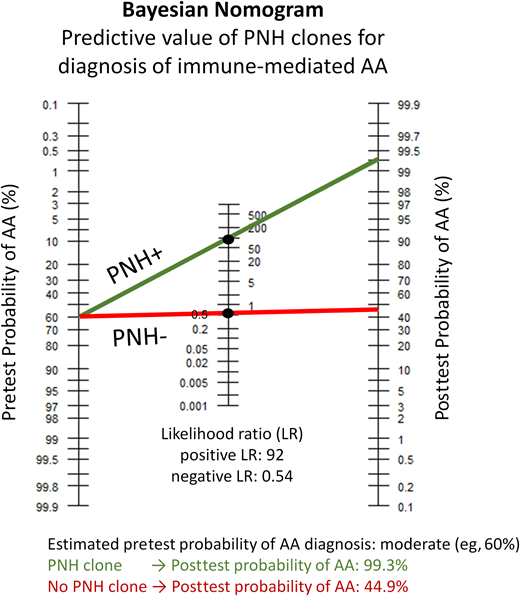
The Bayesian (Fagan's) nomogram demonstrating the pretest and posttest probability of having a diagnosis of immune-mediated AA based on PNH flow cytometry results. PNH clones have a near 100% positive predictive value for the diagnosis of immune-mediated AA, with a positive LR of approximately 92 for immune-mediated AA.8 Shown is an example corresponding to Case 2 in the text. To apply the nomogram tool, one first has to estimate the pretest probability of AA based on the clinical presentation. This previously healthy patient developed pancytopenia in young adulthood without any physical examination findings or family history suggestive of a congenital marrow failure disorder. Based on this presentation, her pretest probability of immune-mediated acquired AA is estimated as moderate to high. The green line shows the results for a conservative estimate of a moderate (approximately 60%) pretest probability of AA, crossing at the positive LR of 92. Detection of a PNH clone increases the posttest probability of AA to 99.3%, allowing to confidently establish the diagnosis of immune-mediated AA and initiate appropriate therapy. Importantly, a negative PNH test would not significantly change the pretest probability. Starting with a pretest probability of 60%, a negative PNH test (shown by the red line crossing at the negative LR of 0.54) will result in a posttest probability of 44.9%. The calculations for the Bayesian nomogram plot are based on Shah et al.8
The Bayesian (Fagan's) nomogram demonstrating the pretest and posttest probability of having a diagnosis of immune-mediated AA based on PNH flow cytometry results. PNH clones have a near 100% positive predictive value for the diagnosis of immune-mediated AA, with a positive LR of approximately 92 for immune-mediated AA.8 Shown is an example corresponding to Case 2 in the text. To apply the nomogram tool, one first has to estimate the pretest probability of AA based on the clinical presentation. This previously healthy patient developed pancytopenia in young adulthood without any physical examination findings or family history suggestive of a congenital marrow failure disorder. Based on this presentation, her pretest probability of immune-mediated acquired AA is estimated as moderate to high. The green line shows the results for a conservative estimate of a moderate (approximately 60%) pretest probability of AA, crossing at the positive LR of 92. Detection of a PNH clone increases the posttest probability of AA to 99.3%, allowing to confidently establish the diagnosis of immune-mediated AA and initiate appropriate therapy. Importantly, a negative PNH test would not significantly change the pretest probability. Starting with a pretest probability of 60%, a negative PNH test (shown by the red line crossing at the negative LR of 0.54) will result in a posttest probability of 44.9%. The calculations for the Bayesian nomogram plot are based on Shah et al.8
Because PNH clones can emerge later in the disease course, repeating PNH flow cytometry can help to identify newly emergent clones in patients who may have tested negative at diagnosis.23 PNH clones found later in the disease course may be particularly helpful by affirming the immune-mediated etiology of AA in patients with refractory AA, in whom the diagnosis of immune-mediated AA may be in question.
In most studies of AA outcomes, PNH clones, regardless of clone size, were associated with improved response to immunosuppressive therapy and better overall prognosis.9,13,29-34 The reasons for improved responses to immunosuppression in patients with PNH clones are incompletely understood but may be partially due to a more accurate diagnosis of immune-mediated AA. PNH clones are also associated with a lower rate of MDS progression.9,31,34
When does a PNH clone require therapy?
CLINICAL CASE 3 (CLASSICAL PNH)
A 20-year-old male college athlete sought treatment from the emergency room 3 times over the past 6 months with acute onset of nausea, vomiting, and abdominal pain, each time resolving with hydration and supportive care. Contrast-enhanced abdominal computed tomography revealed an edematous mesentery with prominent mesenteric lymph nodes without other pathology. Liver function studies showed indirect hyperbilirubinemia of 2.7 mg/dL and an elevated aspartate aminotransferase of 81 U/mL. A complete blood count showed WBCs of 12 × 103 cells/µL, hemoglobin of 12.0 g/dL, and platelets of 109 × 103 cells/µL, which downtrended to WBCs of 4 × 103 cells/µL, hemoglobin of 10.3 g/dL, and platelets of 95 × 103 cells/µL after intravenous hydration. LDH was elevated at596 IU/L (3.10 × ULN), haptoglobin was undetectable, and absolute reticulocytes were 112 × 103 cells/µL. The Coombs test was negative, and the D-dimer was elevated to 3.87 µg/mL. Upper extremity superficial thrombophlebitis was noted at an old intravenous line placement site. Flow cytometry revealed a PNH clone with 78.6% PNH granulocytes, 87.3% PNH monocytes, and 0.8% type II and 11.2% type III PNH RBCs. The patient recalled multiple episodes of darker-colored urine. A bone marrow biopsy, performed to assess for underlying bone marrow failure, showed a 40% cellular marrow without dysplastic features, a normal karyotype, and a disease-associated somatic BCOR mutation. The patient was diagnosed with classical PNH, which likely arose from subclinical nonsevere AA. He was started on folic acid and initiated low-dose aspirin for management of thrombophlebitis and general thromboprophylaxis. Following vaccination for Neisseria meningitides, he started eculizumab therapy with full symptom resolution. Once ravulizumab became available, he was switched to ravulizumab for patient convenience. Three years after the initial diagnosis, PNH flow cytometry showed an expansion of his PNH clone to 92.8% in granulocytes, 94.1% in monocytes, and 36.5% in RBCs (3.4% type II and 33.1% type III). Laboratory studies performed before the maintenance ravulizumab infusion showed WBCs of 3.9 × 103 cells/µL, hemoglobin of 13.0 g/dL, and platelets of 162 × 103 cells/µL, with reticulocytes of 217 × 103 cells/µL and LDH of 282 U/L (1.47 × ULN).
The decision to initiate complement inhibitor therapy in a patient with a PNH clone depends on multiple factors, including clone size, clinical hemolysis, and symptom burden (Table 1). Patients whose primary clinical picture is that of bone marrow failure on average have smaller PNH clones, have little in the way of PNH symptoms, and rarely benefit from PNH-directed therapy. The anemia, frequently seen in patients with AA, most commonly stems from the underlying bone marrow failure and is not caused by hemolysis. Thus, a careful evaluation of the etiology of anemia with particular attention to adequate reticulocyte response is essential when deciding on the utility of adding complement inhibition to standard AA therapy.
In contrast, complement inhibitors improve outcomes in patients with classical PNH,35 who have overt hemolysis, are frequently highly symptomatic, and invariably have large (generally >50%12,14 ) PNH clones. Nearly one-third of patients with classical PNH have abdominal pain.36 Other classical PNH symptoms include fatigue, dyspnea, esophageal spasm, erectile dysfunction, neurologic dysfunction, and gallstone disease. Before the availability of complement inhibitors, 37% to 39% of patients with classical PNH had thrombotic events, frequently involving unusual vascular beds (eg, mesenteric and cerebrovascular), which were the leading cause of mortality.36-38 Less recognized serious complications of PNH include subclinical pulmonary emboli and pulmonary hypertension,39 cerebrovascular ischemia,40 and renal dysfunction.41
Risk factors for thrombosis in patients with PNH clones include granulocyte clone size more than 50%, LDH 1.5 or more times the ULN, high PNH symptom burden, and previous thrombotic events (Table 3).36-38,42-44 When evaluating LDH, it is important to compare a patient's LDH result with the reference range from the testing laboratory, as normal LDH values vary between testing sites due to variations in testing methodologies and platforms. RBC clones more than 3% to 5% and granulocyte clones over approximately 23% are predictive of elevated LDH, with each 10% rise in granulocyte clone size associated with approximately 1.6 higher odds of thrombosis.37 Although PNH clone size correlates closely with hemolysis and LDH,45 the relationship between clone size, hemolysis, and thrombotic risk is not always linear. Rare patients with more than 50% PNH granulocytes have developed thrombotic events despite a near-normal or normal LDH.47 Conversely, patients with PNH clones smaller than 50% can be at increased risk of thrombosis.44,45 Although hypomorphic PIGA mutations cause only partial GPI-anchored protein deficiency (type II cells), which could be less susceptible to hemolysis, thromboses in patients with type II PNH-predominant clones have been reported.47
Risk factors for thrombotic complications in PNH
| Study | Year | Population | Study design | No. of patients | PNH testing method | Risk factor for thrombosis | Effect of PNH clone size |
|---|---|---|---|---|---|---|---|
| de Latour et al36 | 2008 | Patients treated in 58 hematologic centers in France | Retrospective | 460 | Ham test or flow cytometry (PNH clones >5%) | • Age >55 years: HR 1.8 • Transfusions: HR 1.7 • Thrombosis at diagnosis: HR 3.7 • Warfarin as primary prophylaxis: HR 5.2 | >50% clones associated with higher risk of TE: HR 3.2 |
| Lee et al44 | 2013 | South Korean National PNH Registry | Retrospective | 301 | Ham test, sucrose-lysis test, and flow cytometry (no minimal cutoff) | • LDH ≥1.5 × ULN: OR 7.0 Higher predictive value in combination with symptoms: • LDH ≥1.5 × ULN plus abdominal pain: OR 17.8 • LDH ≥1.5 × ULN plus chest pain: OR 19.0 • LDH ≥1.5 × ULN plus dyspnea: OR 10.3 • LDH ≥1.5 × ULN plus hemoglobinuria: OR 10.3 | No significant effect of clone size PNH clone <20%: 16% TE PNH clone 20%-50%: 19% TE PNH clone >50%: 20% TE |
| Schrezenmeier et al45 | 2014 | International PNH Registry | Retrospective | 900 | Clinical diagnosis and/or flow cytometry (no minimal cutoff) | • LDH ≥1.5 × ULN: 15.6% (vs 8.4% <1.5 × ULN) • PNH clone ≥50% | PNH clone <10%: 5.3% TE PNH clone 10%-49%: 7.7% TE PNH clone ≥50%: 15.4% TE |
| Long et al46 | 2017 | Peking Union Medical College Hospital | Retrospective | 104 | Flow cytometry (PNH clones >1%) | • PNH clone ≥50%: OR 9.78 • ABO gene rs495828 GT+TT: OR 5.63 • ABO gene rs2519093 TC+TT: 5.95 | PNH clone ≥50%: OR 9.78 |
| Griffin et al. 201947 | 2019 | Leeds UK PNH Database | Retrospective study of thrombosis in patients with PNH with PNH clone >10% and LDH <2 × ULN) | 25 of 429 patients had PNH clone >10% and LDH <2 × ULN | Flow cytometry (PNH clones >10%) | • Group 1: PNH white cells >30%, PNH red cells <10%, LDH <2 × ULN: 6 of 11 (54%) had TE • Group 2: PNH white cells >30%, PNH red cells >10% with higher proportion of type II red cells than type III red cells, LDH <2 × ULN: 2 of 11 (18%) had TE • Group 3: PNH white cells 10%-30%, PNH red cells <10%, LDH <2 × ULN (0 of 3 had thrombosis) | In patients with TEs, PNH clone ranged from 49% to 100%; median clone sizes were 93% (group 1) and 89% (group 2) |
| Huang et al.48 | 2019 | Peking Union Medical College Hospital | Retrospective | 99 | Flow cytometry (PNH clones >1%) | • PNH clone ≥80%: OR 1.056 • Hemoglobin ≤7.5 g/dL: OR 4.202 • Platelets >100 × 109/L: OR 6.547 • ABO gene rs495828 = G: OR 5.243 | Only evaluated cutoff of PNH clone ≥80%: OR 1.056 |
| Hoechsmann et al43 [abstract] | 2020 | International PNH Registry | Retrospective case control | 57 TE cases, 189 non-TE controls | NA | • Recent HDA: OR 2.65 • LDH ≥1.5 × ULN plus 2-3 HDA symptoms: OR 8.61 • LDH ≥1.5 × ULN plus ≥4 HDA symptoms: OR 14.5 • History of TE: OR 3.6 • History of MAVE: OR 2.17 • Recent prophylactic anticoagulation: OR 4.35 | PNH clone ≥50% |
| Füreder et al49 | 2020 | Austrian PNH network | Retrospective | 59 | Flow cytometry | 14 of 59 patients had TE; 5 of 14 patients had clone size recorded, which ranged from 80% to 100% | Larger clone size in patients with TE |
| Study | Year | Population | Study design | No. of patients | PNH testing method | Risk factor for thrombosis | Effect of PNH clone size |
|---|---|---|---|---|---|---|---|
| de Latour et al36 | 2008 | Patients treated in 58 hematologic centers in France | Retrospective | 460 | Ham test or flow cytometry (PNH clones >5%) | • Age >55 years: HR 1.8 • Transfusions: HR 1.7 • Thrombosis at diagnosis: HR 3.7 • Warfarin as primary prophylaxis: HR 5.2 | >50% clones associated with higher risk of TE: HR 3.2 |
| Lee et al44 | 2013 | South Korean National PNH Registry | Retrospective | 301 | Ham test, sucrose-lysis test, and flow cytometry (no minimal cutoff) | • LDH ≥1.5 × ULN: OR 7.0 Higher predictive value in combination with symptoms: • LDH ≥1.5 × ULN plus abdominal pain: OR 17.8 • LDH ≥1.5 × ULN plus chest pain: OR 19.0 • LDH ≥1.5 × ULN plus dyspnea: OR 10.3 • LDH ≥1.5 × ULN plus hemoglobinuria: OR 10.3 | No significant effect of clone size PNH clone <20%: 16% TE PNH clone 20%-50%: 19% TE PNH clone >50%: 20% TE |
| Schrezenmeier et al45 | 2014 | International PNH Registry | Retrospective | 900 | Clinical diagnosis and/or flow cytometry (no minimal cutoff) | • LDH ≥1.5 × ULN: 15.6% (vs 8.4% <1.5 × ULN) • PNH clone ≥50% | PNH clone <10%: 5.3% TE PNH clone 10%-49%: 7.7% TE PNH clone ≥50%: 15.4% TE |
| Long et al46 | 2017 | Peking Union Medical College Hospital | Retrospective | 104 | Flow cytometry (PNH clones >1%) | • PNH clone ≥50%: OR 9.78 • ABO gene rs495828 GT+TT: OR 5.63 • ABO gene rs2519093 TC+TT: 5.95 | PNH clone ≥50%: OR 9.78 |
| Griffin et al. 201947 | 2019 | Leeds UK PNH Database | Retrospective study of thrombosis in patients with PNH with PNH clone >10% and LDH <2 × ULN) | 25 of 429 patients had PNH clone >10% and LDH <2 × ULN | Flow cytometry (PNH clones >10%) | • Group 1: PNH white cells >30%, PNH red cells <10%, LDH <2 × ULN: 6 of 11 (54%) had TE • Group 2: PNH white cells >30%, PNH red cells >10% with higher proportion of type II red cells than type III red cells, LDH <2 × ULN: 2 of 11 (18%) had TE • Group 3: PNH white cells 10%-30%, PNH red cells <10%, LDH <2 × ULN (0 of 3 had thrombosis) | In patients with TEs, PNH clone ranged from 49% to 100%; median clone sizes were 93% (group 1) and 89% (group 2) |
| Huang et al.48 | 2019 | Peking Union Medical College Hospital | Retrospective | 99 | Flow cytometry (PNH clones >1%) | • PNH clone ≥80%: OR 1.056 • Hemoglobin ≤7.5 g/dL: OR 4.202 • Platelets >100 × 109/L: OR 6.547 • ABO gene rs495828 = G: OR 5.243 | Only evaluated cutoff of PNH clone ≥80%: OR 1.056 |
| Hoechsmann et al43 [abstract] | 2020 | International PNH Registry | Retrospective case control | 57 TE cases, 189 non-TE controls | NA | • Recent HDA: OR 2.65 • LDH ≥1.5 × ULN plus 2-3 HDA symptoms: OR 8.61 • LDH ≥1.5 × ULN plus ≥4 HDA symptoms: OR 14.5 • History of TE: OR 3.6 • History of MAVE: OR 2.17 • Recent prophylactic anticoagulation: OR 4.35 | PNH clone ≥50% |
| Füreder et al49 | 2020 | Austrian PNH network | Retrospective | 59 | Flow cytometry | 14 of 59 patients had TE; 5 of 14 patients had clone size recorded, which ranged from 80% to 100% | Larger clone size in patients with TE |
HDA, recent high disease activity, defined as occurring within 6 months of TE event, LDH ≥1.5 × ULN, and hemoglobin <10 g/dL or at least 1 of the following symptoms: abdominal pain, dyspnea, dysphagia, fatigue, hemoglobinuria, and male erectile dysfunction; HR, hazard ratio; MAVE, major adverse vascular event; OR, odds ratio; TE, thromboembolic event.
In Case 3, the patient has near-normal hemoglobin and notably does not require therapy due to anemia. However, he is at high risk of thrombotic complications. His risk factors for thrombosis include a large PNH clone more than 50%, overt hemolysis with LDH more than 3 times the ULN, high symptom burden, and previous superficial thrombophlebitis. As evidenced by mesenteric edema and elevated D-dimer, his abdominal pain is likely caused by microvascular ischemia in the mesenteric vasculature. He requires prompt initiation of a complement inhibitor to prevent life-threatening thrombotic complications.
The prothrombotic state in PNH is managed most effectively by complement inhibition, which reduces thrombotic events from approximately 7.4% to 1% per patient-year.50 The mechanism of thrombosis in PNH involves abnormal platelet activation by both complement and hemolytic processes as well as abnormal clot composition.37 Untreated patients with PNH have higher fibrinogen and thrombin generation levels, leading to faster-forming fibrin clots that are harder to break down; these abnormalities are improved by eculizumab.51 Primary anticoagulation prophylaxis has been proposed for patients with more than 50% PNH granulocytes who have no contraindications to anticoagulation.38 However, primary anticoagulation prophylaxis in PNH remains controversial. Patients with PNH who have concomitant thrombocytopenia have increased risks of bleeding with anticoagulation, which, before the advent of complement inhibitors, have contributed to PNH mortality. In addition, unlike complement inhibitors, therapeutic anticoagulation alone does not entirely prevent thrombosis in patients with PNH.37,38,50 In situations where patients with classical PNH do not have access to complement inhibitors, primary anticoagulation prophylaxis should be considered. After complement inhibitor therapy has been initiated, primary anticoagulation prophylaxis can be discontinued.
As of 2021, the US Food and Drug Administration has now approved 3 complement inhibitors for the treatment of PNH. Eculizumab and the longer-acting ravulizumab both target terminal complement C5 and effectively abrogate intravascular hemolysis, reduce thrombotic events, and improve quality of life of patients with PNH.52-55 However, approximately 4% to 27% of patients treated with C5 inhibitors continue to experience anemia due to extravascular hemolysis from the accumulation of unopposed C3b on PNH RBCs.56 Although C5 inhibition does not address extravascular hemolysis, the recently approved, first-in-class C3 inhibitor pegcetacoplan, which acts more proximally in the complement cascade, was found to be superior to eculizumab in improving hemoglobin and normalizing hemolytic markers.57 Approved for both frontline or second-line therapy of PNH, the twice-weekly, subcutaneously administered agent pegcetacoplan will be particularly useful in patients who remain moderately or severely anemic despite C5 inhibition because of extravascular hemolysis. Several other proximal complement inhibitors, some of which are orally administered, are in clinical trials.58
Summary
PNH remains a very rare, orphan disease with a widely available and highly sensitive screening test, the results of which can have nuanced implications for diagnosis, follow-up, and complement inhibitor therapy. PNH clones are pathognomonic for immune-mediated bone marrow failure. Patients with AA, who comprise most individuals with PNH clones, have smaller PNH clones and are rarely symptomatic from PNH. In the absence of hemolysis or PNH symptoms, patients with AA with subclinical PNH clones do not benefit from complement inhibitors but should be monitored prospectively as most classical PNH arises out of AA. In contrast, complement inhibitors improve outcomes of patients with classical PNH, who invariably have large PNH clones, clinical hemolysis, and a high burden of PNH-associated symptoms. An individualized approach is required when evaluating patients between these extremes, and referral to a tertiary care center with expertise in managing PNH and AA is recommended.
Acknowledgments
The author would like to thank the current and former members of the Penn/CHOP Bone Marrow Failure Center for helpful discussions as well as patients and their families for participating in research studies of bone marrow failure. This work was supported by NHLBI grant K08 HL132101.
Conflict-of-interest disclosure
Daria V. Babushok: declares no competing financial interests.
Off-label drug use
Daria V. Babushok: no off-label drug use discussed.
