

Abstract
After 3 decades of clinical trials, repeated proof-of-concept success has now been demonstrated in hemophilia A and B gene therapy. Current clinical hemophilia gene therapy efforts are largely focused on the use of systemically administered recombinant adeno-associated viral (rAAV) vectors for F8 or F9 gene addition. With multiple ongoing trials, including licensing studies in hemophilia A and B, many are cautiously optimistic that the first AAV vectors will obtain regulatory approval within approximately 1 year. While supported optimism suggests that the goal of gene therapy to alter the paradigm of hemophilia care may soon be realized, a number of outstanding questions have emerged from clinical trial that are in need of answers to harness the full potential of gene therapy for hemophilia patients. This article reviews the use of AAV vector gene addition approaches for hemophilia A and B, focusing specifically on information to review in the process of obtaining informed consent for hemophilia patients prior to clinical trial enrollment or administering a licensed AAV vector.
Learning Objectives
Understand information important to review with hemophilia patients prior to consenting for a clinical trial or approved AAV vector
Differentiate information that is well understand from information that remains incompletely understood
CLINICAL CASE
A 62-year-old man with severe hemophilia A (HA) with a history of an inhibitor, eradicated by immune tolerance induction, on emicizumab prophylaxis is interested in enrolling in a HA gene therapy trial or receiving a licensed adeno-associated viral (AAV) vector that will be available soon. His comorbidities include 3 target joints, a prior 2-decade history of the hepatitis C virus (HCV) infection with stage 2 liver fibrosis, and HIV well controlled on antiretroviral medications. He wants to know what is known and unknown about hemophilia gene therapy.
Introduction: What are the current approaches for hemophilia gene therapy?
Clinical approaches in hemophilia gene therapy nearly exclusively use systemically delivered recombinant AAV (rAAV) vectors to target hepatocyte expression of an episomally maintained transgene expressed under a hepatocyte promoter (Table 1). The vector is engineered from naturally occurring AAV, which is nonpathogenic in humans and replication defective. rAAV vectors are produced in cell culture using either HEK293 mammalian cells or baclovirus/Sl9 insect cell culture. The design and manufacturing of rAAV vectors was recently reviewed by Li and Samulski.1
Ongoing hemophilia A and B gene therapy trials
| Sponsor | Clinicaltrials.gov | Manufacturing platform | Transgene | Capsid serotype | Dose (vg/kg) | Phase | Ref |
|---|---|---|---|---|---|---|---|
| HB | |||||||
| UCL/St. Jude | NCT00979238 | Mammalian | scFIX | AAV8 | 2 × 1011 to 2 × 1012 | 1/2 | 16,45 |
| Pfizer/Spark | NCT03681273 NCT03307980 | Mammalian | ssFIX-R338L | SPK100 | 5 × 1011 | 1/2, 3 | 9 |
| uniQure | NCT02396342 | Sl9 (insect) | ssFIX-R338L | AAV5 | 2 × 1013 | 3 | 8 |
| Freeline | NCT03369444 | Mammalian | scFIX-R338L | AAVS3 | 7.5 × 1011 9.5 × 1011 | 10 | |
| HA | |||||||
| BioMarin | NCT02576795 NCT03392974 NCT03370913 | Sl9 (insect) | ssFVIII-SQ | AAV5 | 6 × 1013 | 1/2, 3 | 11,15 |
| Sangamo/Pfizer | NCT03061201 | Sl9 (insect) | ssFVIII-SQ | AAV6 | 3 × 1013 | 1/2, 3 | 18 |
| Spark | NCT03003533 NCT03432520 | Mammalian | ssFVIII-SQ | LK03 | 5 × 1011 to 2 × 1012 | 1/2 | 14 |
| UCL/St. Jude | NCT03001830 | Mammalian | ssFVIII-V3 | AAV8 | 6 × 1011 to 6 × 1012 | 1/2 | 4,5 |
| Bayer/Ultragenyx | NCT03588299 | Mammalian | ssFVIII-SQ | AAVhu37 | 5 × 1012 to 2 × 1013 | 1/2 | 58 |
| Takeda | NCT03370172 | Mammalian | ssFVIII-SQ | AAV8 | 1/2 | ||
| Sponsor | Clinicaltrials.gov | Manufacturing platform | Transgene | Capsid serotype | Dose (vg/kg) | Phase | Ref |
|---|---|---|---|---|---|---|---|
| HB | |||||||
| UCL/St. Jude | NCT00979238 | Mammalian | scFIX | AAV8 | 2 × 1011 to 2 × 1012 | 1/2 | 16,45 |
| Pfizer/Spark | NCT03681273 NCT03307980 | Mammalian | ssFIX-R338L | SPK100 | 5 × 1011 | 1/2, 3 | 9 |
| uniQure | NCT02396342 | Sl9 (insect) | ssFIX-R338L | AAV5 | 2 × 1013 | 3 | 8 |
| Freeline | NCT03369444 | Mammalian | scFIX-R338L | AAVS3 | 7.5 × 1011 9.5 × 1011 | 10 | |
| HA | |||||||
| BioMarin | NCT02576795 NCT03392974 NCT03370913 | Sl9 (insect) | ssFVIII-SQ | AAV5 | 6 × 1013 | 1/2, 3 | 11,15 |
| Sangamo/Pfizer | NCT03061201 | Sl9 (insect) | ssFVIII-SQ | AAV6 | 3 × 1013 | 1/2, 3 | 18 |
| Spark | NCT03003533 NCT03432520 | Mammalian | ssFVIII-SQ | LK03 | 5 × 1011 to 2 × 1012 | 1/2 | 14 |
| UCL/St. Jude | NCT03001830 | Mammalian | ssFVIII-V3 | AAV8 | 6 × 1011 to 6 × 1012 | 1/2 | 4,5 |
| Bayer/Ultragenyx | NCT03588299 | Mammalian | ssFVIII-SQ | AAVhu37 | 5 × 1012 to 2 × 1013 | 1/2 | 58 |
| Takeda | NCT03370172 | Mammalian | ssFVIII-SQ | AAV8 | 1/2 | ||
sc, self-complementary; ss, single stranded; UCL, University College London.
While hepatocytes are the natural site of factor IX (FIX) synthesis, liver sinusoidal endothelial cells are the predominate site of factor VIII (FVIII) synthesis.2 Most HA rAAV vectors contain a B-domain deleted FVIII, FVIII-SQ transgene that retains full procoagulant function and accommodates AAV vector packaging constraints (4.7 kB).3 This exact amino acid sequence has been used for recombinant protein HA therapy for 2 decades (eg, Xyntha/ReFacto®, Pfizer) without evidence of increased risk of inhibitor formation. A single trial is using an alternative B-domain deleted FVIII, FVIII-V3 that consists of the SQ linker with 6 additional N-linked glycosylation sites to improve expression.4,5 All enrolling hemophilia B (HB) trials have adapted the use of the FIX-Padua (FIX-R338L) mutation, which is a naturally occurring missense mutation with approximately 8-fold greater specific activity relative to wild-type FIX.6-10
What are the short- and long-term rAAV safety considerations?
In the 300-fold range of rAAV vector doses (2 × 1011 to 6 × 1013 vg/kg) thus far evaluated in hemophilia clinical trials, there have been no major safety concerns.
In the short term, a small number of subjects across 3 trials at rAAV vector doses ranging from 2 × 1012 to 6 × 1013 vg/kg have experienced acute vector infusion reactions, including a single incidence of anaphylaxis and a couple of incidences of either myalgias, fever, and/or hypotension of unclear etiology that may be consistent with an innate immune response to rAAV11-14 ; these events have not clearly correlated with clinical outcomes post vector.
Because AAV efficiently targets the liver, the bulk of safety considerations of systemic rAAV delivery have focused on hepatotoxicity. Within hemophilia trials, a single study outlined multimonth transaminase elevations post vector of unclear etiology that were not associated with liver dysfunction and resolved.11,15 Beyond this, most transaminase elevations observed in hemophilia trials occurred in the setting of an immune response to the rAAV capsid.9,14,16-18 This AAV capsid immune response is hypothesized to occur when CD8+ T cells recognize capsid peptides on the surface of transduced hepatocytes, triggering a cellular immune response that results in clearance of the transduced cells.19 While the AAV capsid immune response has not posed safety concerns in hemophilia, it has diminished or prevented efficacy in some trials.9,16-18 Close monitoring for a capsid immune response within the first 3 months or so post vector classically includes factor assays, liver function studies, and peripheral blood mononuclear cell interferon-γ anticapsid enzyme-linked immunosorbent spot assays that, when interpreted as consistent with a capsid immune response, trigger intervention with immune-modulating therapies to maintain transgene expression; glucocorticoids have been used predominantly,9,16-18 but other immune-modulating agents have been used concurrently with glucocorticoids or in isolation to maintain transgene expression.10,13,14 Additional study is needed to understand which regimen best maintains transgene expression. Further still, because some studies have employed glucocorticoids while simultaneously reporting that participants had no cellular immune response, additional study is needed to distinguish a cellular immune response vs other potential etiologies for elevated transaminases.12,13,15
While safety in hemophilia has been excellent, rAAV vectors developed for other monogenic disorders suggest there may be dose-limiting hepatic toxicities with systemic rAAV vector doses >1 × 1014 vg/kg. Specifically, Zolgensma (Novartis), an rAAV9 vector and the first licensed systemic rAAV vector, delivered at a dose of 1.1 × 1014 vg/kg demonstrated not only remarkable efficacy for spinal muscular atrophy but also evidence of hepatic toxicity in clinical trial such that it was approved with a boxed safety warning for acute liver injury. Subsequently, a case report outlined 2 children presenting 6 to 8 weeks post Zolgensma infusion with acute liver failure that resolved with glucocorticoid intervention.20 While the exact etiology is unclear, the timing post vector, liver biopsy specimens demonstrating CD8+ T-cell infiltration, and responsiveness to steroids suggest an immune-mediated process that is possibly consistent with an rAAV capsid immune response. These observations support the possibility that the rAAV capsid immune response has safety implications and support using the lowest possible rAAV vector dose to minimize safety concerns and maximize efficacy. Additionally, a study using an rAAV8 vector for X-linked myotubular myopathy reported that 3 of 17 boys in its vg/kg dose cohort developed liver failure approximately 6 weeks post vector and ultimately succumbed to complications.21 The gene therapy community eagerly awaits a composite analysis of these observations. Nonetheless, these tragic events support possible dose-limiting toxicities of systemic rAAV vectors at doses >1 × 1014 vg/kg, which are >1.6- to 500-fold higher than the rAAV doses used in hemophilia.
Finally, dorsal root ganglia (DRG) toxicity has recently emerged as a potential short- or long-term safety concern. Overwhelmingly, these observations are histological findings in large-animal models. Specifically, a meta-analysis of NHP data (n = 33 vectors, n = 256 NHPs) by a single laboratory following heterogeneous routes of administration (intrathecal, intracisternal magna, and systemic), vector doses, AAV capsids, and transgenes reported no or mild histological evidence of DRG toxicity in most animals that appeared to be dose dependent and more common with direct central nervous system administration.22 Importantly, clinical symptomatology in only 2 animals was attributed to DRG toxicity. Subsequent NHP studies by the same group suggested DRG toxicity may be related to an unfolded protein response abrogated when inhibiting transgene expression in the DRG with, as yet, unclear clinical translation.23 Thus far, in the full cadre of clinical AAV trials spanning 3 decades, various routes of administration for a variety of disorders, and an approximate 1000-fold range of rAAV vector doses,24 no DRG toxicity clinical symptoms have been observed outside of a single recently reported case. A trial participant who received an rAAVrh10 vector (a capsid not currently used for hemophilia) by intrathecal injection to deliver microRNA for amyotrophic lateral sclerosis presented with symptoms and imaging consistent with DRG toxicity; symptoms diminished with glucocorticoid and supportive care intervention but had not been completely ameliorated at the time of publication.25 Whether DRG toxicity will be route of administration, capsid, or rAAV vector dose dependent and/or a potential rAAV vector platform toxicity is unclear.
The major identified long-term safety concerns of systemic rAAV vectors are the risks of hepatotoxicity and genotoxicity. Direct sequencing data in animals and humans demonstrate that AAV integration can occur and has a propensity for sites of active transcription.26-30 Data in neonatal mice demonstrated AAV integration that disrupted the murine Rian locus (that does not have a human ortholog) and resulted in near 100% penetrance of hepatocellular carcinoma (HCC).29 A subsequent murine study demonstrated that the risk of HCC correlated directly with the rAAV vector dose and degree of cellular division and that hepatocyte-specific enhancer and promoter elements protected against oncogenesis, while non-hepatocyte-specific elements increased HCC risk.27 This would thus support using the lowest possible effective vector dose with hepatocyte-specific promoter and enhancer elements to minimize genotoxicity risk. Recently, studies in HA dogs followed for 10 years post rAAV vector demonstrated evidence of clonal proliferation at sites of AAV integration in liver tissue without tumorigenesis but were the first large-animal data to support the theoretical risk of genotoxicity.30
Nearly all severe hemophilia patients >40 years of age contracted iatrogenic hepatitis B virus and/or HCV, which are known risk factors for HCC such that the hemophilia population has an increased risk of HCC relative to the general population.31 Published 15-year follow-up data of the first participants to receive a systemic rAAV vector (HB subjects) demonstrated no evidence of long-term hepatoxicity, but the report is limited in interpretation due to small cohort size and the absence of detectable transgene expression or direct sequencing data to confirm the persistence of transduced hepatocytes.32 A single clinical trial participant in an HB trial with prior long-term HCV infection and other risk factors for HCC developed HCC approximately 1 year following vector infusion. Recently presented tumor sequencing data demonstrated the expected random, low-frequency AAV integrations without evidence of clonality as well as well-described HCC-associated genetic changes, suggesting that rAAV vector administration did not contribute to HCC development.33 There is thus far no clinical evidence to suggest that systemic rAAV vector delivery is a risk factor for the development of HCC. Ultimately, cohorts outside the adult hemophilia population may more closely mimic the risk factors identified in mice for the development of HCC post systemic rAAV vector infusion. Specifically, infants and toddlers (who have inherent greater degrees of cellular division than adults) who receive higher systemic rAAV vector doses that contain ubiquitous promoters will be important populations to follow to evaluate long-term AAV genotoxicity.
What are the transgene safety considerations?
The generally identified safety concerns related to transgene expression include an immunological response to the expressed protein and direct transgene toxicity. Taking the latter first, epidemiological studies have outlined that supraphysiologic FIX:C has a moderate, independent risk of venous thrombosis (odds ratio, 1.8-4.0) relative to supraphysiologic FVIII:C (OR, 8.8-21.3).34 Biochemical data support that FIX-R338L function, like wild-type FIX, requires FVIIIa cofactor function for enzymatic activity and is similarly activated by FXIa and inactivated by antithrombin (Figure 1), supporting that FIX-R338L is regulated the same as wild-type FIX7 and is not inherently not prothrombotic. Consistent with this, observed thrombotic events in mice expressing wt-FIX or FIX-R338L correlated with the degree of supratherapeutic FIX:C, independent of the expressed transgene.35 Concurrently, HB gene therapy trials using the FIX-R338L transgene achieved FIX:C in the range of mild or normal HB without safety concerns. A single FIX-R338L trial reported thrombosis in a subject who achieved supraphysiologic FIX:C with multiple prothrombotic comorbidities (obesity, kidney failure with an arteriovenous fistula).10 This observation when paired with epidemiological data of supraphysiologic factor activity and venous thrombosis risk underscores the importance of maintaining expression within a therapeutic window. In addition to thrombosis risk, the expression of FVIII or FIX-R388L could impart direct cellular toxicity, which is one possible cause of the unexpected declining FVIII expression observed in the first HA gene therapy trial detailed below.
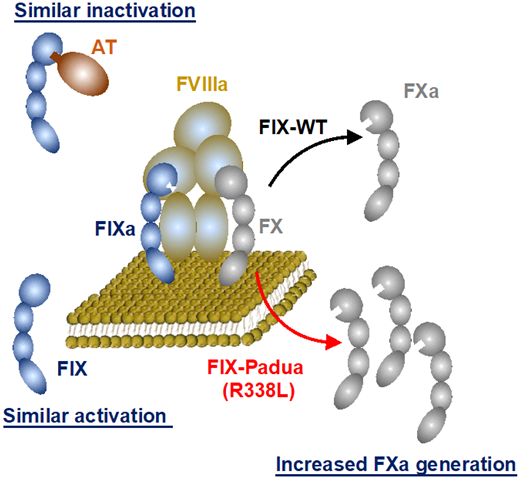
Biochemical characterization of FIX-Padua (FIX-R338L). Data support that FIX-R338L requires factor VIIIa (FVIIIa) cofactor function for enzymatic activity to generate factor Xa (FXa) from FX and is similarly activated for factor XIa (FIXa) and inactivated by antithrombin (AT). Modified with permission from Samelson- Jones et al.7
Biochemical characterization of FIX-Padua (FIX-R338L). Data support that FIX-R338L requires factor VIIIa (FVIIIa) cofactor function for enzymatic activity to generate factor Xa (FXa) from FX and is similarly activated for factor XIa (FIXa) and inactivated by antithrombin (AT). Modified with permission from Samelson- Jones et al.7
While multiple immunological responses to the expressed transgene are possible, the most concerning is the development of inhibitory antibodies, which have not been observed in clinical trial. However, current trial enrollment includes patients who are the least likely to develop an inhibitor (eg, participants with prior or current inhibitors are excluded and must have >50-150 factor exposures). Nonetheless, inhibitor risk post gene therapy is generally thought to be unlikely because of small- and large-animal data that demonstrate the ability of hepatocyte-directed gene therapy to induce tolerance to FVIII, FIX, or FIX-R338L.35-37 These animal studies show that future gene transfer may ultimately be an effective means for FVIII tolerance induction in HA patients with inhibitors; initiating clinical trials for tolerance induction was recently supported by consensus recommendations following a National Heart, Lung, and Blood Institute State of the Science of FVIII inhibitors.38
What are the preliminary efficacy data?
The approximate homogeneous and remarkable phenotypic amelioration that has been observed with heterogeneous transgene-derived factor activity supporting factor activity itself should not be the sole distinguishing feature of clinical programs (Figure 2). Specifically, currently available data are consistent with HA natural history studies that suggest factor activity >10% ameliorates spontaneous bleeding; however, higher factor activity is likely required among patients with marked preexisting joint disease. Most trials report an unpredictable range of expression of 5- to 10-fold variability (some trials up to 50- to 100-fold) such that it is not currently possible to target expression based on individual patient needs.9,10,11,13,14,39 Correspondingly, an up to 7-fold variability in transduction was demonstrated in a chimeric murine model of human hepatocytes.40 This degree of variable expression in a clonal mouse population is consistent with the multiple variables that exist from vector infusion to transgene expression1 that may necessitate tolerance for a range of transgene expression, albeit hopefully to a lesser degree than has thus far been reported. This underscores the need for an accurate measurement of in vivo activity of transgene hemostatic function to define the range of tolerable safe and efficacious expression.

Aggregated reported annualized bleeding rate data for hemophilia A and B clinical trials before (blue) and after (maroon) receiving the described recombinant AAV vectors. The dotted line denotes an annualized bleeding rate of 1.AAV8-wt-FIX, also known as scAAV2/8-LP1-hFIXco. Data from Nathwani et al.45 AMT-060 data from Miesbach et al.12 AMT-061, now etranocogene dezaparvovec, data from Pipe et al.8 SPK-9001, now fidancogene elaparvovec, data from George et al.9 BMN270, now valoctocogene roxaparvovec, data from Pasi et al.11 SPK-8011 data from George et al.14
Aggregated reported annualized bleeding rate data for hemophilia A and B clinical trials before (blue) and after (maroon) receiving the described recombinant AAV vectors. The dotted line denotes an annualized bleeding rate of 1.AAV8-wt-FIX, also known as scAAV2/8-LP1-hFIXco. Data from Nathwani et al.45 AMT-060 data from Miesbach et al.12 AMT-061, now etranocogene dezaparvovec, data from Pipe et al.8 SPK-9001, now fidancogene elaparvovec, data from George et al.9 BMN270, now valoctocogene roxaparvovec, data from Pasi et al.11 SPK-8011 data from George et al.14
In HA and HB efforts, there is a discrepancy in transgene- derived factor activity by one-state assay (OSA) and chromogenic assay (CSA). Transgene-derived FVIII:C by OSA measures approximately 1.6-fold higher than CSA in humans15,39-42 and mice,43 which differs from the experience with recombinant protein products of the same amino acid sequence. Which assay correlates with in vivo hemostatic benefit is unclear. Current trials target exogenous FVIII expression, and whether this mildly alters biochemical properties of the protein is hypothesized (eg, changes in von Willebrand factor affinity or FVIII cleavage by thrombin or factor Xa) but not determined. Similarly, FIX-R338L determined that FIX:C measures higher by OSA vs CSA FIX:C; however, unlike HA, this observation is also maintained with recombinant FIX-R338L and consistent with the available biochemical understanding of FIX-R338L.44 Additionally, unlike HA trials, OSA determined that FIX:C from FIX-R338L expression differs by initiating reagents.44
How long will the therapy last?
Thus far, clinical trial efforts in HB gene transfer mirror hemophilia canine models of rAAV-mediated gene transfer that demonstrated no decline in FVIII or FIX expression for up to 8 years post gene transfer.16,30,42,45,46 In contrast, the first phase 1/2 HA gene therapy trial of valoctocogene roxaparvovec at a dose of 6 × 1013 vg/kg demonstrated an average loss of approximately 40% of transgene expression from year 1 to year 2 post vector (n = 7)11,15 ; similar observations were observed among the 17 phase 3 participants administered the same vector dose who were followed for >2 years.13 Available phase 1/2 data of participants 5 years post valoctocogene roxaparvovec demonstrated a continued, but lesser, decline in FVIII expression in years 2 to 5.47 In contrast, a phase 1/2 study of SPK-8011 for HA at vector doses 5 × 1011 to 2 × 1012 vg/kg demonstrated different pharmacokinetics of expression such that the 12 participants with sustained expression outside the cellular immune response and followed for >2 years demonstrated no apparent decline in expression from year 1 to year 2 (Figure 3).14 Some of these subjects have been followed for up to 4 years and demonstrate generally stable FVIII expression after 1 year post vector; these preliminary SPK-8011 data generally support the current strategy of targeting hepatocytes for multiyear stable FVIII expression. Given that the transgene is maintained episomally, some degree of declining expression over multiyear follow-up may be anticipated, although it is unclear how rapidly, but the mechanism of loss or the more marked decline in FVIII expression from year 1 to year 2 observed with valoctocogene roxaparvovec is unknown. Possible hypotheses include (1) an unfolded protein response, which has been described in mammalian expression systems of recombinant FVIII production and in mouse models of supratherapeutic but not low levels of FVIII expression post rAAV48-50 ; (2) promoter silencing; (3) an ongoing undetected/unmitigated immune response to AAV or the transgene; or (4) failure to form stable concatermized episomal DNA due to properties of the vector.
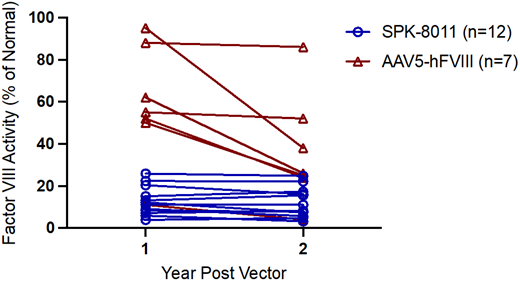
FVIII activity 1 and 2 years post vector infusion in trial participants who received a therapeutic vector dose (6 × 1013 vg/kg of AAV5-hFVIII vs 5 × 1011 to 2 × 1012 vg/kg of SPK 8011) that maintained expression and were followed >2 years post vector. FVIII activity is reported by 1-stage assay for SPK-8011 participants and CSA in participants who received AAV5-hFVIII. Data for AAV5-hFVIII are from Pasi et al11 and data for SPK-8011 are from George et al.14
FVIII activity 1 and 2 years post vector infusion in trial participants who received a therapeutic vector dose (6 × 1013 vg/kg of AAV5-hFVIII vs 5 × 1011 to 2 × 1012 vg/kg of SPK 8011) that maintained expression and were followed >2 years post vector. FVIII activity is reported by 1-stage assay for SPK-8011 participants and CSA in participants who received AAV5-hFVIII. Data for AAV5-hFVIII are from Pasi et al11 and data for SPK-8011 are from George et al.14
What do AAV neutralizing antibodies mean?
AAV neutralizing antibodies (NAb) are incorporated into most eligibility criteria in hemophilia and other disease models using systemic AAV vectors. While results of the assay have an impact on eligibility, the assays are not standardized, can yield highly variable results depending on the assay methods (eg, transduction vs enzyme-linked immunosorbent assay based, employed multiplicity of infection, etc), and do not always have a clear correlation with clinical outcome. Nonetheless, most available data support the fact that high-titer AAV NAb preclude target tissue transduction of a systemically administered rAAV vector and thus limit efficacy. A single AAV5 trial using a dose of 2 × 10 vg/kg is enrolling participants independent of AAV NAb status and has demonstrated transgene expression in all participants except the participant with the highest measured AAV NAb (>1:3000).8,13 These data challenge existing notions that would otherwise predict preexisting AAV NAb to preclude efficacy. Details of the AAV NAb, thus far unreported, are necessary to interpret the data and whether these data are specific to rAAV5 and/or the vector dose used or may be partially explained by methods used to perform the AAV NAb.
Following rAAV vector administration, patients universally develop persistent, multiserotype cross-reactive AAV NAb that available data suggest would preclude repeat rAAV vector efficacy.32,51 This underscores that patients interested in gene therapy must recognize that the current state of clinical development likely only permits 1 systemic rAAV vector infusion. Thus, outlining the current known and unknown information regarding rAAV gene therapy for hemophilia is essential to provide true informed consent for both clinical trials and anticipated soon-to-be licensed vectors (Figure 4); this will be necessary to avoid “buyer's remorse” should one receive an rAAV vector that is ultimately found to be inferior to another rAAV vector.
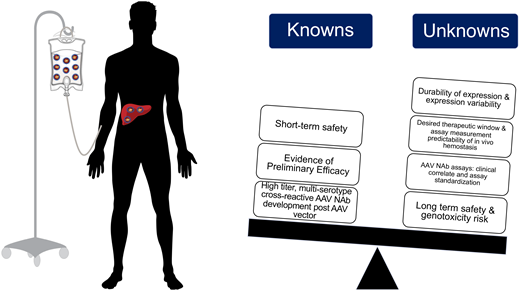
Summary of known and unknown variables for recombinant AAV gene edition clinical efforts for in hemophilia A and B.
Summary of known and unknown variables for recombinant AAV gene edition clinical efforts for in hemophilia A and B.
What other approaches are in development?
Safely achieving sustained, stable, and predictable FVIII or FIX expression, even in the presence of an immune response, is an unrealized goal of hemophilia gene therapy. Beyond gene addition approaches, gene editing using lipid nanoparticles to deliver mRNA encoding Cas9 and gRNA and a donor FIX cDNA template via an rAAV vector to knockin F9 into the albumen locus have demonstrated normal levels of FIX expression in an NHP model.52 In addition, lentiviral vectors for either systemic infusion,53 ex vivo transduction of hematopoietic stem cells,54-56 or induced pluripotent stem cells are being pursued in preclinical investigation and early-phase clinical trial (clinicaltrials.gov; NCT03818763).57
Will hemophilia gene therapy ever be mainstream?
Building on the remarkable successes the field has seen over the past approximately 5 years will be predicated on the thoughtful investigation of questions that have emerged from clinical trials. Optimism in the field will continue to progress, buoyed by 3 general strengths: (1) a strong understanding of the molecular and biochemical basis of HA and HB, (2) an organized patient advocacy and physician provider network that fosters basic research and clinical trials, and (3) a rapidly expanding knowledge base in gene therapy with significant financial investment. Collectively, these strengths ensure that gene therapy will fulfill its promise to alter the paradigm of hemophilia care.
Acknowledgments
This work was supported by NIH/NHLBI K08 HL 146991 (L. A. George).
Dr. George thanks Benjamin Samelson-Jones, MD/PhD, for his thoughtful review of the article.
Conflict-of-interest disclosure
Lindsey A. George: scientific advisory board member: STRM.Bio; data safety monitoring committee member: Avrobio; consultancy: Intellia, Biomarin, Pfizer, Bayer.
Off-label drug use
Lindsey A. George: glucocorticoids were discussed.
