

Abstract
Inherited bone marrow failure syndromes (IBMFS) cause hematopoietic stem progenitor cell (HSPC) failure due to germline mutations. Germline mutations influence the number and fitness of HSPC by various mechanisms, for example, abnormal ribosome biogenesis in Shwachman-Diamond syndrome and Diamond-Blackfan anemia, unresolved DNA cross-links in Fanconi anemia, neutrophil maturation arrest in severe congenital neutropenia, and telomere shortening in short telomere syndrome. To compensate for HSPC attrition, HSPCs are under increased replication stress to meet the need for mature blood cells. Somatic alterations that provide full or partial recovery of functional deficit implicated in IBMFS can confer a growth advantage. This review discusses results of recent genomic studies and illustrates our new understanding of mechanisms of clonal evolution in IBMFS.
Learning Objectives
Learn recent advances in our understanding of clonal hematopoiesis in IBMFS
Recognize early signs of malignant transformation in IBMFS
Introduction
Inherited bone marrow failure syndromes (IBMFS) manifest as ineffective and stressed hematopoiesis due to germline mutations that cause hematopoietic stem progenitor cell (HSPC) failure. The causes of HSPC failure are different depending on disease mechanisms: unresolved DNA cross-links in Fanconi anemia (FA), telomere shortening in dyskeratosis congenita (DC) (also known as short telomere syndromes [STSs]), and ribosomopathy in Shwachman-Diamond syndrome (SDS) and Diamond-Blackfan anemia (DBA).1 However, clinical consequences are similar in that patients experience ineffective hematopoiesis, leading to replicative stress and HSPC exhaustion, and frequently develop malignant transformation (Table 1).
Inherited BMF syndromes
| Syndrome | Causative genes (inheritance) | Etiology | Clinical manifestations | Frequent chromosomal abnormalities | Frequent somatic mutations |
|---|---|---|---|---|---|
| SDS | SBDS (AR)—m/c DNAJC21 (AR) SRP54 (AD) ELF1 (AR) | Ribosomopathy | Hematopoietic/oncologic: neutropenia, anemia, thrombocytopenia, aplastic anemia, MDS, AML Nonhematologic: exocrine pancreatic insufficiency, failure to thrive, malabsorption, short stature, skeletal abnormalities (chondrodysplasia or congenital thoracic dystrophy), endocrine abnormalities, liver dysfunction, cognitive impairment | 20q– (interstitial deletion q11.21-q13.32)—loss of EIF6 i(7)(q10)—duplication of hypomorphic SBDS allele High risk: –7/7q– Complex | EIF6—improve ribosome biogenesis and protein translation efficiency High risk: Biallelic TP53—impaired cell cycle checkpoint and apoptosis |
| FA | FANCA (AR)—m/c FANCB (XR) FANCC (AR) FANCD1/BRCA2 (AR) FANCD2 (AR) FANCE (AR) FANCF (AR) FANCG (AR) FANCI (AR) FANCJ/BRIP1/BACH1 (AR) FANCL (AR) FANCM (AR) FANCN/PALB2 (AR) FANCO/RAD51C (AR) FANCP/SLX4 (AR) FANCQ/ERCC4 (AR) FANCR/RAD51 (AD) FANCS/BRCA1 (AR) FANCT/UBE2T (AR) FANCU/XRCC2 (AR) FANCV/REV7 (AR) FANCW/RFWD3 (AR) | Inability to repair DNA interstrand cross-links | Hematologic/oncologic: thrombocytopenia, anemia, neutropenia, aplastic anemia, MDS, leukemia, squamous cell carcinoma (head and neck, anogenital, esophageal), embryonal tumors (medulloblastoma, neuroblastoma, Wilms tumor in patients with biallelic BRCA2 mutations) Nonhematopoietic: short stature, low birth weight, failure to thrive, microcephaly, microphthalmia, hearing loss, triangular face, micrognathia, cardiac abnormalities (patent ductus arteriosus, atrial septal defect, ventricular septal defect), tracheoesophageal fistula, esophageal atresia, horseshoe or ectopic kidneys, thumb/radius abnormalities, hypoplastic thenar eminence, clinodactyly, café-au-lait spots, hypo/hyperpigmentation of the skin | 1q+ High risk: 3q+ –7/7q– Complex Cryptic RUNX1 translocation | Somatic reversion of FANC genes by back mutation, intragenic recombination, second site mutations suppressing the effect of the original mutation, frame-restoring second site insertions and deletions High risk: RUNX1 RAS pathway (KRAS, PTPN11)—(small number of cases) |
| SCN | ELANE (AD)—m/c HAX1 (AR) GFI1 (AD) G6PC3 (AR) JAGN1 (AR) TCIRG1 (AD) WAS (XR) CSF3R (AD, AR) | Neutrophil maturation arrest and apoptosis | Hematologic/oncologic: severe neutropenia at birth, recurrent infections, MDS, AML | High risk: –7/7q– Complex | CSF3R—enhanced and prolonged response to G-CSF High risk: RUNX1 |
| Short telomere syndrome | TERT (AD, AR)—m/c TERC (AD) DKC1 (XR) NOLA3/NOP10 (AR) NOLA2/NHP2 (AR) TINF2 (AD) WRAP53/TCAB1 (AR) CTC1 (AR) RTEL1 (AD, AR) ACD/TPP1 (AD, AR) PARN (AD, AR) NAF1 (AD) STN1 (AD) NPM1 (AD) ZCCHC8 (AD) | Telomere shortening | Hematologic/oncologic: thrombocytopenia, anemia, neutropenia, pancytopenia, MDS, AML, immunodeficiency, squamous cell carcinoma Nonhematopoietic: the mucocutaneous triad (reticulated skin pigmentation, nail dystrophy, oral leukoplakia), short stature, low birth weight, failure to thrive, hearing loss, retinopathy, mucosal strictures, pulmonary fibrosis, liver fibrosis | High risk: –7/7q– Complex | Somatic reversion— back mutation of DKC1, TERT promoter GOF mutation, mitotic recombination High risk: TP53a |
| DBA | RPS19 (AD)—m/c RPL3 (AD) RPL5 (AD) RPL7 (AD) RPL11 (AD) RPL14 (AD) RPL15 (AD) RPL18 (AD) RPL19 (AD) RPL23 (AD) RPL26 (AD) RPL27 (AD) RPL31 (AD) RPL35a (AD) RPL36 (AD) PRS7 (AD) RPS8 (AD) RPS10 (AD) RPS15 (AD) RPS17 (AD) RPS24 (AD) RPS26 (AD) RPS27 (AD) RPS27A (AD) RPS28 (AD) RPS29 (AD) GATA1 (XR) TSR2 (XR) | Ribosomopathy; red blood cell aplasia | Hematologic/oncologic: Anemia, reticulocytopenia, MDS, AML, solid tumors (osteogenic sarcoma, lung, colon and cervix) Nonhematologic: low birth weight, growth retardation, developmental delay, short stature, microcephaly, other craniofacial malformation, congenital glaucoma or cataract, strabismus, short neck, thumb abnormalities, horseshoe kidney, hypospadias, cardiac abnormalities | Abnormal cytogenetics infrequent | TP53,aPPM1D, ASXL1 (small number of cases)b |
| SAMD9/SAMD9L disorders | SAMD9 (AD) SAMD9L (AD) | Growth inhibition | MIRAGE syndrome Hematologic/oncologic: Thrombocytopenia, anemia, MDS with monosomy 7, recurrent infections, bleeding Nonhematologic: intrauterine growth restriction, developmental delay, adrenal hypoplasia, chronic diarrhea, genital anomalies Ataxia-pancytopenia syndrome Hematologic/oncologic: Pancytopenia, anemia, MDS with monosomy 7/del(7q), AML, recurrent infections Nonhematologic: ataxia, cerebellar atrophy, retinal dysfunction, behavioral abnormalities, alveolar proteinosis | –7/7q– 5q– | Somatic reversion by LOF SAMD9 or SAMD9L mutations ETV6, RUNX1, SETBP1 (small number of cases) |
| Syndrome | Causative genes (inheritance) | Etiology | Clinical manifestations | Frequent chromosomal abnormalities | Frequent somatic mutations |
|---|---|---|---|---|---|
| SDS | SBDS (AR)—m/c DNAJC21 (AR) SRP54 (AD) ELF1 (AR) | Ribosomopathy | Hematopoietic/oncologic: neutropenia, anemia, thrombocytopenia, aplastic anemia, MDS, AML Nonhematologic: exocrine pancreatic insufficiency, failure to thrive, malabsorption, short stature, skeletal abnormalities (chondrodysplasia or congenital thoracic dystrophy), endocrine abnormalities, liver dysfunction, cognitive impairment | 20q– (interstitial deletion q11.21-q13.32)—loss of EIF6 i(7)(q10)—duplication of hypomorphic SBDS allele High risk: –7/7q– Complex | EIF6—improve ribosome biogenesis and protein translation efficiency High risk: Biallelic TP53—impaired cell cycle checkpoint and apoptosis |
| FA | FANCA (AR)—m/c FANCB (XR) FANCC (AR) FANCD1/BRCA2 (AR) FANCD2 (AR) FANCE (AR) FANCF (AR) FANCG (AR) FANCI (AR) FANCJ/BRIP1/BACH1 (AR) FANCL (AR) FANCM (AR) FANCN/PALB2 (AR) FANCO/RAD51C (AR) FANCP/SLX4 (AR) FANCQ/ERCC4 (AR) FANCR/RAD51 (AD) FANCS/BRCA1 (AR) FANCT/UBE2T (AR) FANCU/XRCC2 (AR) FANCV/REV7 (AR) FANCW/RFWD3 (AR) | Inability to repair DNA interstrand cross-links | Hematologic/oncologic: thrombocytopenia, anemia, neutropenia, aplastic anemia, MDS, leukemia, squamous cell carcinoma (head and neck, anogenital, esophageal), embryonal tumors (medulloblastoma, neuroblastoma, Wilms tumor in patients with biallelic BRCA2 mutations) Nonhematopoietic: short stature, low birth weight, failure to thrive, microcephaly, microphthalmia, hearing loss, triangular face, micrognathia, cardiac abnormalities (patent ductus arteriosus, atrial septal defect, ventricular septal defect), tracheoesophageal fistula, esophageal atresia, horseshoe or ectopic kidneys, thumb/radius abnormalities, hypoplastic thenar eminence, clinodactyly, café-au-lait spots, hypo/hyperpigmentation of the skin | 1q+ High risk: 3q+ –7/7q– Complex Cryptic RUNX1 translocation | Somatic reversion of FANC genes by back mutation, intragenic recombination, second site mutations suppressing the effect of the original mutation, frame-restoring second site insertions and deletions High risk: RUNX1 RAS pathway (KRAS, PTPN11)—(small number of cases) |
| SCN | ELANE (AD)—m/c HAX1 (AR) GFI1 (AD) G6PC3 (AR) JAGN1 (AR) TCIRG1 (AD) WAS (XR) CSF3R (AD, AR) | Neutrophil maturation arrest and apoptosis | Hematologic/oncologic: severe neutropenia at birth, recurrent infections, MDS, AML | High risk: –7/7q– Complex | CSF3R—enhanced and prolonged response to G-CSF High risk: RUNX1 |
| Short telomere syndrome | TERT (AD, AR)—m/c TERC (AD) DKC1 (XR) NOLA3/NOP10 (AR) NOLA2/NHP2 (AR) TINF2 (AD) WRAP53/TCAB1 (AR) CTC1 (AR) RTEL1 (AD, AR) ACD/TPP1 (AD, AR) PARN (AD, AR) NAF1 (AD) STN1 (AD) NPM1 (AD) ZCCHC8 (AD) | Telomere shortening | Hematologic/oncologic: thrombocytopenia, anemia, neutropenia, pancytopenia, MDS, AML, immunodeficiency, squamous cell carcinoma Nonhematopoietic: the mucocutaneous triad (reticulated skin pigmentation, nail dystrophy, oral leukoplakia), short stature, low birth weight, failure to thrive, hearing loss, retinopathy, mucosal strictures, pulmonary fibrosis, liver fibrosis | High risk: –7/7q– Complex | Somatic reversion— back mutation of DKC1, TERT promoter GOF mutation, mitotic recombination High risk: TP53a |
| DBA | RPS19 (AD)—m/c RPL3 (AD) RPL5 (AD) RPL7 (AD) RPL11 (AD) RPL14 (AD) RPL15 (AD) RPL18 (AD) RPL19 (AD) RPL23 (AD) RPL26 (AD) RPL27 (AD) RPL31 (AD) RPL35a (AD) RPL36 (AD) PRS7 (AD) RPS8 (AD) RPS10 (AD) RPS15 (AD) RPS17 (AD) RPS24 (AD) RPS26 (AD) RPS27 (AD) RPS27A (AD) RPS28 (AD) RPS29 (AD) GATA1 (XR) TSR2 (XR) | Ribosomopathy; red blood cell aplasia | Hematologic/oncologic: Anemia, reticulocytopenia, MDS, AML, solid tumors (osteogenic sarcoma, lung, colon and cervix) Nonhematologic: low birth weight, growth retardation, developmental delay, short stature, microcephaly, other craniofacial malformation, congenital glaucoma or cataract, strabismus, short neck, thumb abnormalities, horseshoe kidney, hypospadias, cardiac abnormalities | Abnormal cytogenetics infrequent | TP53,aPPM1D, ASXL1 (small number of cases)b |
| SAMD9/SAMD9L disorders | SAMD9 (AD) SAMD9L (AD) | Growth inhibition | MIRAGE syndrome Hematologic/oncologic: Thrombocytopenia, anemia, MDS with monosomy 7, recurrent infections, bleeding Nonhematologic: intrauterine growth restriction, developmental delay, adrenal hypoplasia, chronic diarrhea, genital anomalies Ataxia-pancytopenia syndrome Hematologic/oncologic: Pancytopenia, anemia, MDS with monosomy 7/del(7q), AML, recurrent infections Nonhematologic: ataxia, cerebellar atrophy, retinal dysfunction, behavioral abnormalities, alveolar proteinosis | –7/7q– 5q– | Somatic reversion by LOF SAMD9 or SAMD9L mutations ETV6, RUNX1, SETBP1 (small number of cases) |
The differential effects of biallelic vs heterozygous TP53 mutations to leukemic transformation have not been studied in IBMFS other than SDS.
Whether somatic mutations contribute to malignant transformation in DBA has not been fully investigated.
m/c, most common; AD, autosomal dominant; AR, autosomal recessive; GOF, gain of function; XR, X-linked recessive.
Successful hematopoietic stem cell transplant (HSCT) can cure bone marrow failure (BMF) and prevent leukemic transformation; however, transplant outcome is poor in patients who have already developed malignant clones before HSCT.2,3 Besides, HSCT comes with a price of potential end-organ damage and accelerated solid tumorigenesis. Therefore, the major goal of surveillance is to detect early signs of malignant transformation and determine the ideal timing for transplant. Although there are no consensus surveillance guidelines with a high level of evidence due to a lack of randomized controlled trials, experts generally agree with serial monitoring of blood counts and bone marrow (BM). Risk-based surveillance recommendations are summarized in the Figure 1.
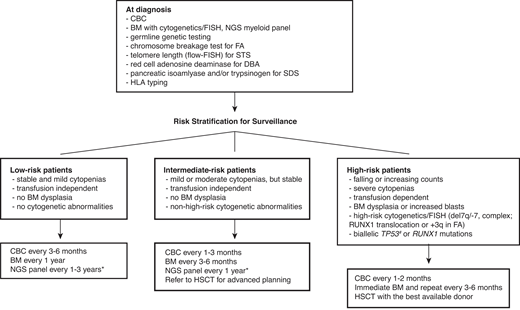
Surveillance recommendations for patients with IBMFS based on the risk of malignant transformation. Detailed diagnostic workup for BMF is described by DeZern and Churpek.7 Once diagnosed, patients are followed by CBC with differential, BM aspiration and biopsy with cytogenetics, FISH, and NGS myeloid panel at regular intervals. The frequency of follow-up depends on the disease and mutation types, baseline counts and BM findings, and the presence of interval changes on subsequent visits. Although the NGS myeloid panel is not a part of standard practice for the management of IBMFS, it is becoming more widely used with advancement of our understanding in CH in both IBMFS and the general population. NGS myeloid panel results are particularly useful to select patients who are at high risk of malignant transformation without overt clinical signs of disease progression. We can increase the frequency of surveillance and initiate discussion and planning for early intervention for the best possible outcome in these high-risk patients. *The optimal frequency and the best genomic source (PB vs BM) of NGS myeloid panel testing have not been fully established. #Although biallelic TP53 mutations are known high-risk features, conventional NGS techniques do not readily provide allelic status of a given mutation. The severity of BMF is defined as the following: mild—ANC <1500/µL, hemoglobin ≥8 g/dL, or platelets 50-150 K/µL; moderate—ANC <1000/µL, hemoglobin <8 g/dL, or platelets <50 K/µL; severe—ANC <500/µL, hemoglobin <8 g/dL, or platelets <30 K/µL. ANC, absolute neutrophil count; CBC, complete blood count; FISH, fluorescence in situ hybridization; PB, peripheral blood.
Surveillance recommendations for patients with IBMFS based on the risk of malignant transformation. Detailed diagnostic workup for BMF is described by DeZern and Churpek.7 Once diagnosed, patients are followed by CBC with differential, BM aspiration and biopsy with cytogenetics, FISH, and NGS myeloid panel at regular intervals. The frequency of follow-up depends on the disease and mutation types, baseline counts and BM findings, and the presence of interval changes on subsequent visits. Although the NGS myeloid panel is not a part of standard practice for the management of IBMFS, it is becoming more widely used with advancement of our understanding in CH in both IBMFS and the general population. NGS myeloid panel results are particularly useful to select patients who are at high risk of malignant transformation without overt clinical signs of disease progression. We can increase the frequency of surveillance and initiate discussion and planning for early intervention for the best possible outcome in these high-risk patients. *The optimal frequency and the best genomic source (PB vs BM) of NGS myeloid panel testing have not been fully established. #Although biallelic TP53 mutations are known high-risk features, conventional NGS techniques do not readily provide allelic status of a given mutation. The severity of BMF is defined as the following: mild—ANC <1500/µL, hemoglobin ≥8 g/dL, or platelets 50-150 K/µL; moderate—ANC <1000/µL, hemoglobin <8 g/dL, or platelets <50 K/µL; severe—ANC <500/µL, hemoglobin <8 g/dL, or platelets <30 K/µL. ANC, absolute neutrophil count; CBC, complete blood count; FISH, fluorescence in situ hybridization; PB, peripheral blood.
Clonal hematopoiesis (CH) is frequently seen in older individuals. Ten percent to 20% of individuals older than 70 years carry somatic mutations in blood, most frequently in genes involved in epigenetic regulation (DNMT3A, TET2, and ASXL1), which is associated with about 1% per year risk for progression to myeloid malignancies.4 Clonal hematopoiesis in IBMFS has a couple of important differences compared with CH observed in older, otherwise healthy individuals; it occurs more frequently and early in life. The mutational spectrum is also quite different. This is likely because preexisting molecular and cellular defects due to germline mutations cause stem cell exhaustion at baseline, and somatic mutations that can compensate underlying defects confer a growth advantage.5 It is currently not established whether the detection of clones with particular mutations or progressive expansion of such clones warrants empirical treatments before frank cytopenias or myelodysplastic syndromes (MDS)/acute myeloid leukemia (AML) arise. For example, CSF3R mutations in severe congenital neutropenia (SCN) or heterozygous TP53 mutations in SDS can persist for years without progression to malignancy, even in the setting of multiple mutations in a given patient. Determination of ideal timing for HSCT in the setting of CH in IBMFS requires further studies.
Somatic reversion is the acquired alteration of inherited pathogenic mutation, resulting in the partial or full functional recovery of the affected protein. It has been reported in many inherited genetic disorders, and potential mechanisms have been reported as back mutation, intragenic recombination, second site mutations suppressing the effect of the original mutation, frame-restoring second site insertions, and deletions.6 It can contribute to CH by conferring survival benefit to reverted clones, but the expansion of hematopoietic clones is not always associated with higher propensity for leukemic transformation. This may be due to the overall improved fitness of HSPC by correcting underlying functional defects while keeping the cell cycle checkpoint intact, most importantly p53, which can actively suppress tumorigenesis.
In this review, the term CH will be used to describe a phenomenon where any HSPC clone with somatic genomic alterations—including single-nucleotide variants, indels, large deletions, or gross chromosomal abnormalities—contributes to a larger pool of mature blood cells than what is expected for any single HSPC clone under homeostasis.
CLINICAL CASE
A 21-year-old man with SDS presented to clinic for a 6-month follow-up. The patient has mild neutropenia and mild thrombocytopenia, and blood counts have been stable in the past 6 months. The BM is hypocellular with mild dysmorphologies in the myeloid lineage, but no frank dysplasia is seen and blasts are not increased. Cytogenetics showed del(20q) in 2 of 20 cells. Peripheral blood myeloid neoplasm panel sequencing result reveals 2 mutations in the EIF6 gene: D112N (variant allelic frequency [VAF] 5%) and N106S (VAF 2%) and 2 mutations in the TP53 gene: R248W (VAF 1%) and R110L (VAF 0.4%). The hematologist decides to monitor his counts every 3 months, as well as BM every 3 to 6 months, and repeat blood myeloid neoplasm panel sequencing in 1 year to monitor the TP53 mutant clones.
Shwachman-Diamond syndrome
SDS is characterized by congenital anomalies; BMF, with neutropenia being the most prominent feature; and exocrine pancreatic insufficiency.8 Biallelic mutations in the SBDS gene are most common (>90%). Less frequently, mutations in DNAJC21, SRP54, and EFL1 present with clinical features of SDS. About 20% to 30% of patients develop MDS/AML, which is frequently accompanied by complex karyotype.2,9-11
A recent sequencing study shed light on our understanding of the mechanisms of clonal evolution in SDS. Clonal hematopoiesis was common in patients with SDS (72% among those without frank MDS/AML).12 Interestingly, mutated genes in SDS were different from those of the general population. Mutations in EIF6 were the most common (59%), followed by TP53 (39.8%) and CSNK1A1 (7.2%), whereas mutations in DNMT3A, TET2, or ASXL1 were less frequent. The most commonly observed chromosomal abnormality was also del(20q) (47%), which is the location for the EIF6 gene. In another study, CH was observed in 59% of patients, and TP53 mutations were the most frequent (48%) (of note, EIF6 was not included in the gene panel in this study).13 The prevalence of TP53 mutations increased with age; about 80% of patients older than 10 years had at least one TP53 mutation, and frequently two or more TP53 mutations were discovered in a given patient. The presence of TP53 mutations among patients with SDS is not an impending sign of leukemic transformation, even though TP53 mutations are frequently encountered in MDS/AML and portend a poor prognosis.
The SBDS protein cooperates with the GTPase EFL1 to release EIF6 from the nascent 60S ribosomal subunit in the cytosol, so that the 60S subunit is free to associate with the 40S subunit and form the mature 80S ribosome.14 Therefore, the loss-of-function (LOF) EIF6 variants would result in increased availability of the 60S subunit for maturation and compensate for the defective SBDS. In fact, EIF6 variants were frequently found in patients with SDS, which was associated with clonal expansion due to improved translational efficiency and stem cell fitness. While functional compensation for primary ribosomal defects improved survival of HSPC in SDS, it did not necessarily increase the risk of malignant transformation. On the other hand, biallelic TP53 mutations were almost always observed in patients with leukemic transformation. P53 is a transcription factor that mediates cell cycle arrest and apoptosis in response to cellular stress signals. Biallelic LOF mutations in TP53 allow HSPCs to progress through cell cycle and escape apoptosis even in the presence of critical cellular stresses posed by ribosomopathy, thereby conferring a growth advantage. Single-cell DNA sequencing on serial samples from patients who developed AML revealed that patients can have multiple independent TP53 mutant clones for a long time, and the particular clone carrying biallelic mutations can give rise to leukemia over several years.12 The presence of biallelic mutations usually cannot be assessed in the conventional bulk sequencing, which only informs VAF but not allelic status of a given mutation in cells. This raises a question whether performing single-cell DNA sequencing regularly on patients with known TP53 mutant clones can help select patients with biallelic TP53 mutations who are at risk of leukemic transformation. Of course, this expensive monitoring approach will need to be carefully assessed in clinical trials to confirm efficacy and cost-effectiveness.
DBA is another inherited ribosomopathy due to heterozygous mutations in the components of either the small 40S or large 60S ribosomal subunits.8 It is characterized by red blood cell aplasia and congenital anomaly. The risk of MDS/AML is relatively low compared with other IBMFS discussed here, with a cumulative incidence of AML of 5% by the age of 46 years.15 The mutational spectrum leading to MDS/AML has not been systematically or functionally studied in DBA. Somatic mutations in TP53 and PPM1D in MDS have been reported in sporadic case reports,16-18 which raises a possibility that similar mechanisms may drive malignant transformation in DBA and SDS.
Fanconi anemia
FA is the most common IBMFS. At least 22 genes have been identified (FANCA-FANCW).19,20 The underlying cellular defect is the inability to repair DNA cross-links, leading to apoptosis via the p53 pathway, or transformation if p53 is inactivated.21 Patients have congenital anomalies, BMF, and cancer predisposition, most notably squamous cell carcinoma of the head and neck.20
Patients have a 30% to 40% cumulative risk of myeloid malignancies by age 40 years.9,22 Myeloid malignancies often arise from clones with abnormal karyotypes, characteristically unbalanced translocations leading to gains or losses of chromosomes. Gain of chromosome 1q is the most common karyotypic abnormality in FA, which can be seen without MDS/AML and does not indicate impending transformation.23 Conversely, monosomy 7/del(7q) are signs of impending transformation, if not transformed already, which requires urgent HSCT. Gain of chromosome 3q, which is associated with increased expression of EVI1, is also highly characteristic in FA and frequently precedes monosomy 7/del(7q).24-26 RUNX1 abnormalities, including cryptic translocation, also indicate high-risk of transformation.23
Single-nucleotide polymorphism array analysis is one of the noninvasive monitoring methods that can identify chromosomal abnormalities from blood samples. From 130 patients with FA (mean age, 14.5 years; range, 0-50 years), single-nucleotide polymorphism array detected chromosomal mosaic events (CMEs) in 16 patients (12.3%).27 Nine of 16 patients carried multiple CMEs. The presence of CMEs was associated with higher incidence of hematologic and solid cancers, as well as increased risk of death.
Large-scale next generation sequencing (NGS) studies have not been published in FA; therefore, it is difficult to speculate the contribution of somatic mutations to clonal evolution. Prior studies using Sanger sequencing of selected oncogenes showed largely negative results, but that may be due to the small number of genes included in the panel and Sanger sequencing not being sensitive enough.23
More recently, targeted myeloid panel sequencing performed on 16 patients with FA with MDS/AML revealed 10 patients had detectable somatic mutations, most frequently involving RUNX1, and the RAS pathway genes (KRAS and PTPN11).28 Of these 16 patients, 12 patients had either chromosome 3 or 7 abnormalities and/or RUNX1 mutations, confirming the prior reports. Another NGS study of 7 patients with FA (including 1 with AML) also confirmed frequent CH from a young age, virtually observed in all patients tested, regardless of the presence of peripheral blood cytopenia or leukemia.29 One AML sample in that study carried multiple MDS/AML mutations, including TP53. Although a clear pattern of clonal evolution cannot be determined from these studies due to small sample sizes, a relative lack of DNMT3A, TET2, and SF3B1 was observed in both studies, suggesting the different somatic mutational spectrum in FA.
Approximately 15% to 18% of patients with FA are reported to have somatic reversion. It frequently results in CH, because recovery of the protein function often confers a growth advantage. It can result in improved counts and stable hematologic course, with longer survival, lower risk of MDS/AML, and less likelihood to require HSCT.30-32 However, malignant transformation in coexisting nonrevertant FA cells may still occur, requiring regular monitoring.30
Severe congenital neutropenia
SCN is characterized by severe neutropenia from birth, complicated by recurrent infections. Treatment with granulocyte colony- stimulating factor (G-CSF) can increase the circulating neutrophil counts and is effective for prevention of recurrent infections. The most common cause is the ELANE gene mutations (∼50%), followed by HAX1 and G6PC3 (10%-20%).13
About 20% of patients taking G-CSF develop MDS/AML in their lifetime.33,34 Somatic mutations in CSF3R (encoding the G-CSF receptor) and RUNX1 have been frequently observed in those who developed MDS/AML.35 From a targeted sequencing study of 40 patients, CH was frequently observed (62%; mean age, 16.6 years).13 As expected, the most frequently observed mutation was the CSF3R mutation (40%).13 All CSF3R variants were nonsense mutations resulting in truncation of cytoplasmic domain of the G-CSF receptor, leading to prolonged and hyperactive signaling in response to G-CSF. Therefore, HSPCs carrying CSF3R mutations have a growth advantage in the presence of G-CSF. Independent CSF3R-mutant clones can coexist, expand over many years, or sometimes disappear.
Another very frequently observed mutation in MDS/AML was the RUNX1 mutation (64.5%), and in fact, approximately 80% of them also had the concurrent CSF3R mutations.33 Serial evaluation of a patient who developed AML with CSF3R and RUNX1 mutations suggests that the CSF3R mutations were likely initiating events conferring a proliferative advantage, and an additional RUNX1 mutation later in the disease progression led to a block in differentiation and fueled leukemogenesis.
Short telomere syndrome
Telomere length is regulated by cooperation of telomerases and telomeric protein complexes (shelterin and CST [CTC1-STN1-TEN1]).36 Germline mutations in telomere components may result in short telomere length, which is associated with BMF, hepatic and pulmonary fibrosis, and cancer predisposition.37 The telomeres are shortened on each cell division due to an “end-replication problem,” where 5′ to 3′ DNA polymerases can only extend the DNA strand in one direction in the presence of a template sequence, leaving the extreme 5′ end of the chromosome as a gap in replication.38 Critically short telomere lengths, below the first percentile for age-matched controls, cause cellular senescence and premature cell death, as well as a clinical syndrome known as STS (also known as DC or telomere biology disorder). Patients exhibit a wide spectrum of disease manifestations, from classical DC with a mucocutaneous triad (oral leukoplakia, lacy reticular skin hyperpigmentation, and nail dystrophy) and early onset BMF to adult-onset MDS/AML, pulmonary fibrosis, or liver disease without apparent BMF.39
All MDS/AML cases in STS were associated with abnormal karyotypes, most commonly monosomy 7 (56%).40 The somatic mutational landscapes in these patients (n = 14; median age 53 years) were similar to that of MDS/AML in the general population. In patients without MDS/AML (median age, 59 years), 6 of 20 evaluated patients had CH (30%) with the mean VAF of 8.2% (range, 2.2%-32.9%). Mutations in TP53 (n = 3) were most commonly observed, followed by TET2 (n = 2).40 On the other hand, 2 other studies with slightly younger individuals reported that somatic mutations associated with MDS/AML were rarely observed, whereas CH was common.41,42 Whether the presence of somatic mutations discovered by NGS is associated with an increased risk of malignant transformation, without other coexisting high-risk clinical features, is not known at this time.
Somatic reversion of the telomerase activity in the peripheral blood has been reported. Examples include acquired gain-of-function mutations in the TERT promoter region in patients with the TERT or PARN heterozygous mutations, leading to overexpression of the wild-type allele.43 A back mutation in DKC142 and mitotic recombination of the TERC gene resulting in isodisomy of the wild-type allele in patients with germline heterozygous TERC mutations have also been reported.44 These somatic reversion events can stabilize the telomere lengths and allow clonal expansion of the reverted clones.
SAMD9/SAMD9L disorders
Germline mutations in SAMD9 and SAMD9L cause MIRAGE (myelodysplasia, infection, restriction of growth, adrenal hypoplasia, genital phenotypes, and enteropathy) syndrome and ataxia pancytopenia syndrome, respectively.45,46 SAMD9 and SAMD9L are paralogs on the chromosome 7q21.2. Both SAMD9 and SAMD9L proteins are endosome fusion facilitators, and they play a role in degradation of growth factor receptors by internalization. Gain-of-function mutations in SAMD9 or SAMD9L have a growth-inhibitory effect by reducing Raf/MEK/ERK signaling, and loss of mutated allele confers growth advantage. Mutant allele is lost frequently by acquired chromosome 7 loss (monosomy 7 or del(7q)) or concomitant somatic LOF mutations.47,48
Conclusion
Clonal hematopoiesis is commonly observed in patients with IBMFS at a much younger age than in the general population. The mutational spectrum is also different in IBMFS: epigenetic regulators, such as DNMT3A, TET2, and ASXL1, the most frequently mutated genes in the aging population, are relatively rare—except in the older patients with STS. Instead, somatic genomic alterations that can restore the original function of protein (eg, somatic reversion of the causative mutation or compensatory second mutations) or that allow pathologic adaptation for survival (eg, by inactivation of p53 or loss of mutant allele) are preferentially selected for clonal expansion (Figure 2). Reduced HSPC numbers and fitness at baseline also create an environment in which clones with such genetic alterations outcompete other HSPCs and quickly establish clonal dominance. However, based on currently available evidence, CH itself, unless accompanied by high-risk cytogenetic abnormalities, progressive cytopenias, and/or BM morphologic changes, does not require immediate definitive treatments even in the setting of IBMFS (Table 1). Future studies should address whether preemptive interventions based on high-risk CH features improve patient outcome in clinical trials.
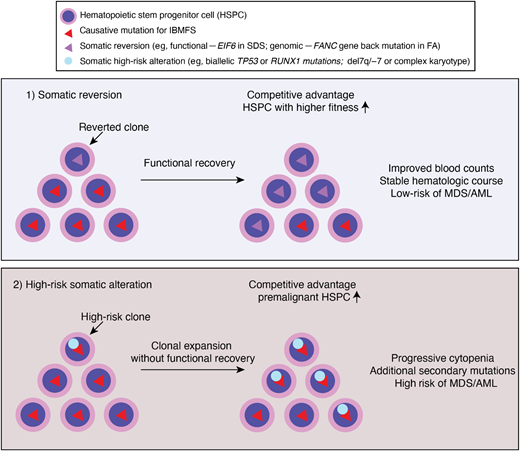
Clonal hematopoiesis in IBMFS. Underlying genetic mutations lead to ineffective and stressed hematopoiesis, and ultimately HSPC attrition. Genetic variations occurred during lifetime of HSPC may confer a growth advantage if it corrected underlying genetic and/or cellular defects by somatic reversion. Alternatively, bypassing cell cycle check point by acquiring high risk genetic features, such as loss of p53, may confer a growth advantage. Clones that reverted underlying defects may have competitive advantage and higher fitness, resulting in improved counts and stable hematologic course. Whereas clones with high risk somatic alterations (eg, TP53 mutations or monosomy 7) may result in clonal expansion without functional recovery of underlying defects. These clones are more prone to acquire additional secondary mutations and/or progress to MDS/AML.
Clonal hematopoiesis in IBMFS. Underlying genetic mutations lead to ineffective and stressed hematopoiesis, and ultimately HSPC attrition. Genetic variations occurred during lifetime of HSPC may confer a growth advantage if it corrected underlying genetic and/or cellular defects by somatic reversion. Alternatively, bypassing cell cycle check point by acquiring high risk genetic features, such as loss of p53, may confer a growth advantage. Clones that reverted underlying defects may have competitive advantage and higher fitness, resulting in improved counts and stable hematologic course. Whereas clones with high risk somatic alterations (eg, TP53 mutations or monosomy 7) may result in clonal expansion without functional recovery of underlying defects. These clones are more prone to acquire additional secondary mutations and/or progress to MDS/AML.
CLINICAL CASE (continued)
This patient carries the 2 most common somatic mutations in SDS, EIF6 and TP53, and these mutations are likely arising from independent clones. Deletion 20q is also a common cytogenetic finding that results in the loss of EIF6. The presence of TP53 mutations is a high-risk feature; however, given the small clone sizes and stable blood counts and BM morphology, impending leukemic transformation is unlikely. Although there is no available consensus guideline, more frequent follow-up with blood counts and BM examination along with serial NGS panel sequencing is desirable for early detection of disease progression.
Acknowledgments
This work was supported in part by National Heart Lung and Blood Institute grant K99 HL150628 (M.J.). Authors thank Dr. Robert Brodsky and Dr. Amy DeZern for their constructive feedback.
Conflict-of-interest disclosure
Haruna Batzorig Choijilsuren: no conflicts to disclose.
Yeji Park: no conflicts to disclose.
Moonjung Jung: no conflicts to disclose.
Off-label drug use
Haruna Batzorig Choijilsuren: nothing to disclose.
Yeji Park: nothing to disclose.
Moonjung Jung: nothing to disclose.
