

Abstract
Rapid advances in sequencing technology have led to the identification of somatic mutations that predispose a significant subset of the aging population to myeloid malignancies. Recently recognized myeloid precursor conditions include clonal hematopoiesis of indeterminate potential (CHIP) and clonal cytopenia of unknown significance (CCUS). These conditions can present diagnostic challenges and produce unwarranted anxiety in some instances. While the risk of progression to myeloid malignancies is very low in CHIP, true CCUS confers an exponential increase in risk. Idiopathic cytopenia of unknown significance (IDUS) lacks the predisposing genetic mutations and has a variable course. In this review we define the early myeloid precursor conditions and their risk of progression. We present our diagnostic approach to patients with unexplained cytopenias and discuss the clinical consequences of CHIP and CCUS.
Learning Objectives
Identify myeloid precursor conditions (CCUS and CHIP) and their clinical consequences
Understand the diagnostic approach to patients with unexplained cytopenias
Understand the role of next-generation sequencing in the workup of unexplained cytopenias
Introduction
Cancer precursor states were first recognized in solid tumors and led to a better understanding of the multistep processes underlying oncogenesis. In hematology, monoclonal gammopathy of undetermined significance and monoclonal B-cell lymphocytosis are well-characterized cancer precursor states. These conditions are defined by the presence of clonally expanded hematopoietic cells, the absence of overt malignancy, and increased risk of developing lymphoid malignancies. In parallel, clonal hematopoiesis and clonal cytopenias are emerging as newly recognized precursor states that increase the risk of developing myeloid malignancies, in particular myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). Rapid advancements in genomic technology have resulted in the detection of cancer-associated mutations without the morphologic changes characteristic of MDS and AML. This has led to the identification of new precursor states with increased predisposition to myeloid malignancies. In this brief review, we discuss the definition, clinical consequences, and management of early myeloid precursor states.
CLINICAL CASE
An 80-year-old man with pancytopenia was referred to our clinic. He had a past medical history of chronic obstructive pulmonary disease and recurrent deep vein thrombosis. His medications included an oral anticoagulant and antihypertensive therapy. He did not report any fatigue, recurrent infections, fever, or night sweats. Initial testing showed the following:
| Hemogram (normal range) | Additional studies (normal range) |
|---|---|
| WBC: 3.16 (4.5-11 K/µL) | Serum iron: 65 (60-150 mcg/dL) |
| RBC: 3.14 (4.5-5.9 M/µL) | Ferritin: 266 (30-400 ng/mL) |
| PLT: 86 (150-350 K/µL) | RDW: 20.7 (11%–14%) |
| Hgb: 10.2 (13-16.3 g/dL) | Absolute reticulocyte count: 43.2 (47-152 K/µL) |
| MCV: 96 (80-100 fL) | TSH, B12, folate, zinc, copper, and lactate dehydrogenase within normal limits Antinuclear antibody negative |
| Hemogram (normal range) | Additional studies (normal range) |
|---|---|
| WBC: 3.16 (4.5-11 K/µL) | Serum iron: 65 (60-150 mcg/dL) |
| RBC: 3.14 (4.5-5.9 M/µL) | Ferritin: 266 (30-400 ng/mL) |
| PLT: 86 (150-350 K/µL) | RDW: 20.7 (11%–14%) |
| Hgb: 10.2 (13-16.3 g/dL) | Absolute reticulocyte count: 43.2 (47-152 K/µL) |
| MCV: 96 (80-100 fL) | TSH, B12, folate, zinc, copper, and lactate dehydrogenase within normal limits Antinuclear antibody negative |
MCV, mean corpuscular volume; RBC, red blood cell; WBC, white blood cell.
Morphologic examination of the bone marrow showed a normocellular marrow with trilineage hematopoiesis and no evidence of dysplasia or increased blasts. Flow cytometry did not show abnormal cells. Conventional karyotyping showed 2 abnormal cell lines: 46,XY,del(12)(q24.1),add(22)(q13)[3]/46,XY,add(10)(q22)[2]/46,XY[18]. Next-generation sequencing (NGS) from his bone marrow revealed the presence of 3 mutations:
TP53 c.743G>A, p.Arg248Gln (NM_000546.5)
Variant allele frequency: 18.6%
U2AF1 c.471G>T, p.Gln157His (NM_006758.2)
Variant allele frequency: 19.7%
DNMT3A c.2173 + 1G>A, p.? (NM_175629.2)
Variant allele frequency: 19.2% Clonal hematopoiesis of indeterminate potential
In 2014, 3 groups of researchers simultaneously discovered the presence of cancer-associated mutations in the peripheral blood of seemingly normal older individuals.1-3 The term clonal hematopoiesis of indeterminate potential (CHIP) was coined and refers to the presence of mutations associated with hematologic malignancies in individuals with normal blood counts and no evidence of hematologic malignancies (Table 1).4 These mutations represent somatic events that lead to the clonal expansion of hematopoietic stem cells. ARCH (age-related clonal hematopoiesis) is another acronym used to describe this phenomenon. The biological mechanisms underlying CHIP are addressed in an accompanying review by L. Gondek.
The most commonly mutated genes in CHIP/CCUS and their respective functions
| Genes | Function/pathway |
|---|---|
| DNMT3A, TET2, ASXL1, IDH1, IDH2 | Epigenetic regulators |
| SF3B1, SRSF2, U2AF1 | RNA splicing |
| TP53, PPM1D | DNA repair/cell cycle |
| JAK2, CBL, GNAS, GNB1 | Signaling |
| BCOR, BCORL1 | Transcription regulation |
| Genes | Function/pathway |
|---|---|
| DNMT3A, TET2, ASXL1, IDH1, IDH2 | Epigenetic regulators |
| SF3B1, SRSF2, U2AF1 | RNA splicing |
| TP53, PPM1D | DNA repair/cell cycle |
| JAK2, CBL, GNAS, GNB1 | Signaling |
| BCOR, BCORL1 | Transcription regulation |
Mutations in DNMT3A, TET2, and ASXL1 are the most frequent mutations in CHIP and confer a lower risk of progression in solitary.
CHIP is relatively common in the aging population, with initial studies estimating a prevalence of roughly 10% among individuals aged 70 to 80. In those studies, CHIP confers a 10-fold increase in the risk of developing a hematologic malignancy.1-3 This translates to a rate of progression of 0.5% to 1% per year, similar to the rate of progression from monoclonal gammopathy of undetermined significance and monoclonal B-cell lymphocytosis to lymphoid malignancies.1-3 Accordingly, most older individuals with CHIP have normal blood counts, no features of hematologic malignancies, and a very low risk of progression. However, the risk of progression from CHIP to myeloid malignancies can vary and depends on several factors. The size of the CHIP clone correlates with the magnitude of risk. A variant allele frequency (VAF) cutoff of 1% to 2% was initially used to define CHIP. This cutoff was based on the lower limit of detection of whole exome sequencing but may not be ideal for current clinical practice. Clones with a VAF of 10% or more demonstrate a 50-fold increase in the risk of developing a hematologic malignancy.1-3 The identity of the mutated genes affects the risk of progression. While genes that code for epigenetic modifiers (DNMT3A, TET2, and ASXL1) are the most commonly mutated in CHIP, they do not confer the highest risk of progression. Two studies examined CHIP in blood samples taken years before patients developed AML and showed a higher prevalence of CHIP in these preleukemic samples.5,6 Mutations in splicing-factor genes (U2AF1, SRSF2, and SF3B1) as well as IDH1, IDH2, and TP53 conferred a higher risk of progression.5,6
The risk of progression is also influenced by the number of mutations in an individual with CHIP. Multiple driver mutations in the same individual are thought to reflect clonal evolution and the acquisition of passenger mutations in the pathway toward frank malignancy.
Blood counts are normal by definition in CHIP, but a higher red cell distribution width (RDW) seems to predict a higher risk of progression.5 A higher RDW is thought to reflect underlying ineffective hematopoiesis and often heralds falling counts and malignant transformation.
Clinical consequences and management of CHIP
Currently, there are no clear clinical indications to screen for CHIP. Nevertheless, due to expanding indications for genetic sequencing, CHIP is increasingly diagnosed in the clinical setting. Current indications for genetic sequencing include germline sequencing for the evaluation of cancer predisposition, for guidance on therapy choices in solid tumors, for detecting circulating tumor DNA, and in the workup of unexplained cytopenias.
In patients with solid tumors and their relatives, testing for germline predisposition is routinely done on DNA extracted from the peripheral blood. Germline predisposition genetic panels can include genes involved in CHIP, such as TP53. Not infrequently, these panels will return results in which mutations are detected at a VAF below the usual heterozygosity rates (VAFs below 40%). These clones could represent CHIP, and additional tissue testing may be needed to clarify this.7,8 Similarly, testing for circulating tumor DNA is done using blood samples and could uncover CHIP clones.9 CHIP clones can also be uncovered when sequencing is conducted using solid-tumor tissue samples due to contamination with blood cells.10
With the increase in CHIP diagnoses, several large centers, including ours, have started dedicated CHIP clinics to evaluate and follow these patients.11 A diagnosis of CHIP in older individuals could produce unwarranted anxiety. The risk of hematologic malignancies for carriers of CHIP who lack high-risk features is minor. It is important to reassure these individuals that most of them will not develop hematologic malignancies and do not require clinical intervention.12 On the other hand, clones with larger VAFs (>10%) and clones with mutations in high-risk genes or with mutations in more than 1 driver gene warrant closer monitoring with serial blood counts. A bone marrow examination should be pursued if these patients develop cytopenias. The workup for cytopenias is further detailed below.
Certain CHIP clones can carry significant risk to patients with solid tumors who will undergo treatment with cytotoxic chemotherapy and/or radiation. Cytotoxic chemotherapy and ionizing radiation are likely to introduce a selective pressure that leads to the expansion of CHIP clones with mutations in the DNA repair pathways (TP53, CHEK2, and PPM1D). The presence of CHIP mutations increases the risk of treatment-related myeloid neoplasms.13 This finding has important clinical implications for patients with solid tumors undergoing adjuvant/neoadjuvant chemotherapy and/or radiation. Currently, no clinical guidelines call for screening for CHIP in this population.
Special attention should also be paid to the cardiovascular risk associated with CHIP. The increase in all-cause mortality detected in earlier CHIP studies was mostly attributable to an increase in fatal cardiovascular events, including ischemic stroke, coronary heart disease, and early-onset myocardial infarction.1,2,14 Mutations in the 2 most common CHIP genes, TET2 and DNMT3A, are associated with an increased risk of cardiovascular disease through proinflammatory mechanisms.1,14 In addition, JAK2 V617F-mutated CHIP is associated with venous thrombosis, including deep vein thrombosis and pulmonary embolism.15 Carriers of CHIP were also found to have worse heart failure outcomes and worse outcomes after aortic valve implantation procedures.12-14 In our CHIP clinic, we assess the presence of classic risk factors of cardiovascular disease, such as hyperlipidemia, diabetes mellitus, hypertension, and smoking. We refer patients who require interventions to our cardio-oncologist for further management.
The diagnostic approach to unexplained cytopenias
Unexplained cytopenias are defined as persistent cytopenias in 1 or more cell lineages without a clear underlying etiology (hemoglobin <13.0 g/dL [males], <12.0 g/dL [females]; absolute neutrophil count <1.8 × 109/L; platelets <150 × 109/L).16 The workup for cytopenias starts with ruling out nutritional, autoimmune, infectious, toxic, and neoplastic causes.17 The term “unexplained cytopenias” is used when the origin of cytopenias is not explained by these causes or by concomitant illnesses.
MDS is one of the most common hematologic malignancies and frequently presents with unexplained cytopenias.17 The workup of unexplained cytopenias starts with ruling out MDS (Figure 1). MDS is a group of heterogeneous clonal hematopoietic malignancies characterized by ineffective hematopoiesis and increased risk of AML. The diagnosis of MDS requires morphological studies of peripheral blood and bone marrow and cytogenetics to identify nonrandom chromosomal abnormalities. Diagnostic criteria for MDS include dysplasia in at least 10% of cells in any hematopoietic lineage, increased myeloblasts, or MDS-defining cytogenetic abnormalities detected through karyotyping.18 It is important to note that morphologic criteria for MDS suffer from great interprovider variability in terms of detecting dysplasia.19
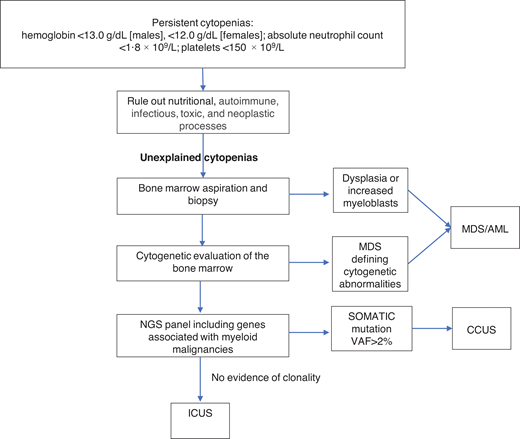
The diagnostic workup for cytopenias starts with ruling out underlying etiologies, including nutritional, autoimmune, infectious, drug-mediated, and neoplastic processes. NGS plays an important role in the workup of unexplained cytopenias to differentiate between CCUS and ICUS.
The diagnostic workup for cytopenias starts with ruling out underlying etiologies, including nutritional, autoimmune, infectious, drug-mediated, and neoplastic processes. NGS plays an important role in the workup of unexplained cytopenias to differentiate between CCUS and ICUS.
NGS plays an important role in the workup of unexplained cytopenias. Although not included in the current diagnostic criteria for MDS, NGS panels from the peripheral blood can aid in the diagnosis of MDS and other hematologic malignancies.20,21 The absence of cancer-associated somatic mutations in the peripheral blood has a high negative predictive value for an underlying myeloid malignancy.22 On the other hand, the presence of these mutations in the peripheral blood raises the suspicion of an underlying hematologic malignancy. The presence of larger clones with VAFs of ≥10% and clones with 2 or more mutations in the peripheral blood has a positive predictive value of 0.86 and 0.88 for the diagnosis of a myeloid neoplasm in cytopenic patients. Conversely, smaller clones involving only 1 mutation in the common epigenetic regulators (DNM3TA, TET2, and ASXL1) have a low predictive value unless a second mutation is present in the same sample. Mutations in spliceosome genes (SF3B1, SRSF2, and U2AF1), JAK2, and RUNX1 have the highest predictive value for myeloid neoplasms irrespective of co-occurring mutations.23
In the absence of diagnostic criteria for MDS, unexplained cytopenia could represent one of two entities depending on the presence of clonal genetic abnormalities: clonal cytopenia of unknown significance (CCUS) or idiopathic cytopenia of unknown significance (ICUS).24-26 It is important to differentiate between CCUS and ICUS because the prognosis and management are different (Table 2). ICUS is defined as unexplained cytopenia that does not meet the minimal criteria for MDS. CCUS is defined as ICUS with one or more somatic mutations typically found in patients with myeloid malignancies. Idiopathic dysplasia of unknown significance is a condition without cytopenia that usually represents a reactive process.24
The unique clinical features differentiating ICUS, CHIP, CCUS, and MDS
| ICUS | CHIP | CCUS | MDS | |
|---|---|---|---|---|
| Cytopenias | + | − | + | + |
| Somatic mutations | − | + | + | + |
| Morphologic dysplasia | − | − | − | + |
| Increased blasts | − | − | − | ± |
| Risk of transformation to AML | Very low | Very low | Low | Low to very high |
| Suggested management and follow-up* | - CBC with differential, history, and physical annually or semiannually | - Routine health maintenance. - Consider annual CBC with differential - Assess cardiovascular risk | - CBC with differential every 3-6 mo - History and physical annually or semiannually - Repeat bone marrow biopsy if counts worsen - Assess cardiovascular risk | Treatment per stage- specific guidelines |
| ICUS | CHIP | CCUS | MDS | |
|---|---|---|---|---|
| Cytopenias | + | − | + | + |
| Somatic mutations | − | + | + | + |
| Morphologic dysplasia | − | − | − | + |
| Increased blasts | − | − | − | ± |
| Risk of transformation to AML | Very low | Very low | Low | Low to very high |
| Suggested management and follow-up* | - CBC with differential, history, and physical annually or semiannually | - Routine health maintenance. - Consider annual CBC with differential - Assess cardiovascular risk | - CBC with differential every 3-6 mo - History and physical annually or semiannually - Repeat bone marrow biopsy if counts worsen - Assess cardiovascular risk | Treatment per stage- specific guidelines |
+, present; −, absent.
Evidence-based guidelines for management of CHIP, CCUS, and idiopathic dysplasia of unknown significance are currently lacking. Follow-up should be tailored to each patient's individual risk and needs.
The clinical significance of CCUS
The current model of progression from CHIP to a myeloid malignancy includes a stepwise progression through a number of disease states. CHIP is the earliest identifiable entity in this model; by definition, it requires the presence of normal blood counts. The development of cytopenia represents the next manifestation of a dysfunctional hematopoietic system progressing toward a frank myeloid malignancy. Cytopenia in CCUS is a result of the clonal hematopoietic process leading to ineffective hematopoiesis. It is important to rule out other causes of cytopenia before diagnosing CCUS. For example, a patient with isolated thrombocytopenia and a low-level DNMT3A mutation could have idiopathic thrombocytopenic purpura (ITP). If the thrombocytopenia responds to treatment with steroids, the patient is unlikely to have CCUS. The DNMT3A mutation in this situation is less significant because DNMT3A-mutated CHIP is common in healthy older individuals.1-3
CCUS confers a much higher risk of progression to a myeloid malignancy than ICUS, with a 10-year cumulative probability of progression of 82% in CCUS vs 9% in ICUS. Furthermore, CCUS clones with a VAF >20% are associated with a more than 95% risk of progression to a myeloid malignancy in a 10-year period.23 Consequently, a VAF of 20% is likely a better cutoff to define CCUS than the 2% used for defining CHIP. The presence of mutations in specific genes (such as U2AF1, ZRSR2, SRSF2, JAK2, or RUNX1) and the presence of multiple mutations also herald a high risk of progression of up to 80% to 90% in 5 years.23 Although the term “CCUS” is now accepted by experts in the field, I would argue that this is a misnomer because of the high risk of progression to myeloid malignancy in true CCUS. Larger clonal events can also be seen in CCUS and are associated with a risk of progression. Trisomy 8 detected by fluorescence in situ hybridization signaled the presence of a low-level clonal process and increased risk of subsequent MDS in a group of patients with unexplained cytopenias.27 Focal copy number aberrations and copy-neutral loss of heterozygosity detected by single-nucleotide polymorphism assays are associated with worse survival in patients with CCUS.28
On the other hand, the clinical course for ICUS is more indolent. Patients with ICUS and no evidence of clonality can be monitored with periodic blood counts. Patients with CCUS require closer monitoring of their blood counts and repeat bone marrow examination if counts worsen (Table 2).
The clone size and the number of mutations in CCUS tend to be much higher than in CHIP. The pattern of mutations in CCUS tends to resemble mutations found in lower-risk MDS.29,30 Furthermore, patients with high-risk CCUS had similar survival to those with myeloid neoplasms.23 High-risk CCUS lies in a close continuum to lower-risk MDS, and the line between these 2 entities continues to blur. Patients with CCUS can also have low-level dysplasia, and some patients may develop transfusion-dependent anemia.29 While no large randomized trials exist in this space, a single-center study examining the treatment of transfusion-dependent CCUS showed some success with MDS-type therapies, including growth factors and hypomethylating agents.31 Patients with high-risk CCUS who develop transfusion dependency or signs and symptoms of bone marrow failure may benefit from treatment algorithms used in low-risk MDS. Consensus treatment and monitoring guidelines for CCUS are currently lacking.
Our patient was diagnosed with high-risk CCUS. Although his peripheral counts were highly suspicious for MDS, he did not have dysplasia, increased myeloblasts, or MDS-defining cytogenetic abnormalities. The patient was counseled about his condition and is being monitored with monthly hemograms due to the high-risk nature of his mutations. A repeat bone marrow examination will be pursued if his counts worsen or if he develops symptoms of bone marrow failure.
Conflict-of-interest disclosure
Afaf E. W. G. Osman: no competing financial interests to declare.
Off-label drug use
Afaf E. W. G. Osman: use of hypomethylating agents and growth factors in CCUS is briefly discussed in this article.
