

Abstract
The treatment of acute graft-versus-host disease (aGVHD) has become more nuanced in recent years with the development of improved risk classification systems and a better understanding of its complex, multisystem pathophysiology. We review contemporary approaches to the risk stratification and initial treatment of aGVHD, including ongoing clinical trials. We summarize the findings that led to the first US Food and Drug Administration approval for steroid-refractory aGVHD (SR-aGVHD), ruxolitinib, as well as some of the challenges clinicians still face in treating SR-aGVHD. Finally, we discuss the evaluation and management of steroid-dependent aGVHD, which affects approximately one-third of patients who have long-term, waxing and waning symptoms distinct from chronic GVHD. Future clinical trials for aGVHD treatment may identify steroid-sparing approaches for patients who have a high likelihood of response and approaches to improve tissue repair and dysbiosis for those unlikely to respond to immunosuppression alone.
Learning Objectives
Identify risk-stratified approaches to the initial management of aGVHD
Identify potential contributing factors for persistent or recurring symptoms after the initial treatment of aGVHD
Introduction
Recent advancements have improved overall survival and decreased the incidence of grades 3 and 4 acute graft-versus-host disease (aGVHD) after allogeneic hematopoietic cell transplantation (HCT), but substantial challenges remain.1-3 High-dose corticosteroids are standard therapy for aGVHD, but this approach does not consider individual factors and may lead to over- or undertreatment. In this review we describe current risk-adapted approaches to aGVHD management, highlighting opportunities for improvement.
Sixty years elapsed between the description of secondary disease as an immunologic complication of murine bone marrow transplantation (1959) and the first US Food and Drug Administration (FDA) approval for SR-aGVHD treatment, ruxolitinib (2019).4,5 With a deeper understanding of the pathophysiology of aGVHD, including a need for tissue repair and reversal of dysbiosis, clinical advancements should be tested at a much faster pace.
CLINICAL CASE
A 47-year-old man with acute myeloid leukemia underwent matched unrelated donor HCT after myeloablative busulfan and cyclophosphamide conditioning with tacrolimus and methotrexate for GVHD prophylaxis. Day 11 methotrexate was 50% dose reduced, and leucovorin rescue was added for severe mucositis. On day 21, he developed a patchy maculopapular skin rash involving a body surface area (BSA) of <20% and was started on 0.1% triamcinolone topical cream. On day 25, he developed fever, nausea and vomiting, and worsening rash involving his face, anterior/posterior torso, and lower extremities to his knees (60% BSA). He had stage 3 skin, stage 1 upper-gastrointestinal (GI), and overall grade 2 Minnesota standard-risk aGVHD.6,7 Treatment included 2 mg/kg/d prednisone and continued tacrolimus. A complete response (CR) by day 28 of therapy was sustained at 8 weeks. Although steroids were discontinued by day 52 of therapy, he had hyperglycemia requiring insulin and became cushingoid during treatment.
Risk-based assessment and initial treatment
Clinical severity-based risk assessment
The grading criteria for aGVHD are well established for the skin, liver, and intestinal tract, with higher grades associated with worse transplant outcomes.7 However, discrepancies have remained in clinical staging across centers, which can influence multicenter trial outcome reporting.8 In order to standardize staging, consensus guidelines were developed at the University of Michigan and subsequently tested in the multicenter Mount Sinai Acute GVHD International Consortium (MAGIC).9,10 These guidelines provide more precise definitions for aGVHD organ staging as well as confidence levels for diagnosis (confirmed, probable, possible, negative) based on supporting evidence (eg, histologic confirmation, clinical action). Experts from the European Society for Blood and Marrow Transplantation, the National Institutes of Health, and the Center for International Blood and Marrow Transplant Research Task Force have recommended the MAGIC criteria for the diagnosis and scoring of aGVHD.11
Simpler classification schemes may further delineate aGVHD risk in order to test personalized treatment strategies in patients with differing organ staging who are likely to have similar outcomes. The Minnesota aGVHD risk score incorporates sites of organ involvement and stage of disease to prognosticate a response to initial aGVHD therapy and the risk of transplant- related mortality.6,12 A web-based calculator is available online to determine the Minnesota aGVHD score (https://redcap.ahc.umn.edu/surveys/?s=bNmFhseJIf). Together, MAGIC staging and the Minnesota score are used in the risk assessment of patients with newly diagnosed aGVHD in many current clinical trials.
Biomarker-based risk assessment
In addition to optimizing clinical GVHD scoring systems, efforts to improve the determination of severity and likelihood of response have focused on identifying peripheral blood biomarkers. Criteria and phases of biomarker development, as well as the different types of biomarkers for acute and chronic GVHD, have recently been reviewed by Adom et al.13 Because some available biomarkers used to diagnose aGVHD are confounded by organ damage or infections, they are of limited use in aGVHD risk assessment.
The MAGIC group identified 2 serum biomarkers of GVHD that in combination predict severe GVHD and nonrelapse mortality (NRM): a suppressor of tumorigenesis, a member of the interleukin 1 receptor family and the soluble receptor for interleukin 33, and regenerating islet-derived 3-alpha, produced by damaged Paneth cells with antimicrobial and epithelial-protective properties.14 In a multicenter validation study, the MAGIC algorithm probability (MAP) and clinical responses were determined at the time of treatment initiation and again after 4 weeks of therapy. Using the initial MAP, patients could be categorized by Ann Arbor (AA) score,1-3 each with a distinct risk of NRM; after 4 weeks, changes in the MAP further refined the estimation of NRM risk in all AA groups. Importantly, the MAP was found to more accurately predict 6-month NRM than a change in clinical symptoms.
Additional biomarkers of inflammation and tissue damage have been investigated. Amphiregulin (AREG), an epidermal growth factor (EGF)-like molecule involved in type 2 immune responses and tissue repair, is another novel biomarker associated with GVHD risk. Elevated levels of AREG have been identified in late aGVHD.15 Additionally, AREG levels have been shown to further refine the Minnesota aGVHD risk score, using samples collected at aGVHD diagnosis as part of the Blood and Marrow Transplant Clinical Trials Network (BMT CTN) 0302 and 0802.16 An AREG threshold of ≥33 pg/mL identified patients with a lower likelihood of day-28 response and a higher 2-year NRM. Pathologically elevated AREG levels decline over time in patients who respond to aGVHD therapy.17 Although biomarker testing is available to clinicians, novel markers may yet be developed, and the role of biomarkers remains an area of active research investigation. The assessment of biomarker-based risk stratification and monitoring should continue in the context of prospective clinical trials.
Risk-adapted initial treatment
For patients with grade 2a manifestations of aGHVD (defined as upper-GI symptoms, stool output <1 L/d, rash <50% BSA, without hepatic involvement), treatment with lower-dose steroids (0.5 mg/kg/d vs 1.0 mg/kg/d) has been shown to be effective without increasing the risk of secondary immunosuppression.18 However, for patients with grade 2b or higher manifestations (defined as stool volume ≥1 L/d, rash ≥50% BSA, or hepatic involvement), treatment with lower-dose steroids (1.0 mg/kg/d vs 2.0 mg/kg/d) was associated with an increased likelihood of requiring secondary immunosuppressive therapy.
Recently, the BMT CTN reported (trial 1501) a randomized phase 2 study testing the steroid-free initial treatment of Minnesota standard risk aGVHD (N = 127) with sirolimus vs prednisone.19 In this study, the day-28 response rate was similar, at 65% for sirolimus (90% CI, 54%-76%) vs 73% (90% CI, 64%-82%) for prednisone. However, patients on the sirolimus arm had fewer side effects from therapy and an improved patient-reported quality of life, suggesting that steroid-free treatment of standard-risk aGVHD is feasible. A note of caution is that <10% of the participants enrolled in BMT CTN 1501 had lower-GI GVHD, and thus we still do not know with confidence how sirolimus therapy would perform as the upfront therapy in this patient population. Additionally, it is unclear how widely these results have been adopted across clinical centers.
Patients with a favorable likelihood of response to aGVHD treatment may be candidates for steroid-sparing approaches; in contrast, those patients at highest risk of fatal aGVHD may be candidates for novel agents. GVHD treatment trials within the last 5 years incorporating risk-assessment algorithms are summarized in Table 1.
First-line risk-adapted treatment trials for aGVHD
| Study number | GVHD risk category | Study type/intervention | Treatment target | Transplant eligibility criteria | Study status | Primary outcome measures |
|---|---|---|---|---|---|---|
| Standard-risk aGVHD | ||||||
| NCT02806947 BMT CTN 1501(19) | Standard-risk (Minnesota risk), previously untreated aGVHD requiring systemic therapy, AA score 1-2 biomarker assessment (performed at randomization) | Randomized, multicenter, phase 2 Sirolimus vs prednisone 2 mg/kg | mTOR inhibitor | Any donor type, conditioning Child, adult, older-adult ages | Completed | Day-28 CR/PR rates similar for sirolimus vs prednisone (64.8% vs 73%) Sirolimus associated with reduced steroid exposure and hyperglycemia, improved QOL, and increased risk for TMA |
| NCT03846479 | Low-risk aGVHD, Minnesota standard risk, AA score 1 | Single arm, monotherapy Itacitinib | JAK1 inhibitor | Any donor type, conditioning Ages 12-75 years | Recruiting | Day-28 Minnesota standard-risk clinical criteria (CR, PR, TRM) Day-28 AA score 1 |
| NCT04144036 | Standard-risk aGVHD, Minnesota risk, AA score 1-2 | Single-center nonrandomized phase 1 Neihulizumab dose escalation | Humanized monoclonal antibody binding to CD162 (PSGL-1) | Any donor type, conditioning Ages ≥18 years | Recruiting | Day-28 SAE, MTD of neihulizumab, plasma Cmax |
| High-risk aGVHD | ||||||
| NCT02133924 | High-risk aGVHD, AA score 3 | Multicenter, phase 2 Natalizumab + prednisone | Humanized recombinant antibody binding to alpha-4 integrin | Any donor type, conditioning Ages ≥18 years | Recruiting | Day-28 CR |
| NCT0252502917 | Arm 1: high-risk aGVHD (Minnesota risk, or AA score 3, or AREG ≥33 pg/mL) Arm 2a: steroid-dependent aGVHD Arm 2b: SR-aGVHD | Nonrandomized phase ½ Standard of care IST + Pregnyl | uhCG, which may promote immune tolerance, provides source of EGF for epithelial repair (Pregnyl®) | Any donor type, conditioning Ages 0-76 years | Active, not recruiting | Phase 1: MTD of uhCG/EGF in combination with standard IST Phase 2: day-28 CR, PR, MR, and no response |
| NCT03158896 | De novo high-risk aGVHD SR-aGVHD | Phase 1, dose escalation Umbilical cord-derived, ex vivo cultured, and expanded Wharton's Jelly MSC | Immunosuppressive properties: suppress proliferation of activated T cells, increase production of regulatory T cells, shift immune response toward tolerance | Ages 18-75 years | Active, not recruiting | Day-45 treatment-related SAE |
| NCT04061876 | High-risk grade 2-4 aGVHD (based on ST2, REG3α, or other experiment object) | Randomized, phase 2 Ruxolitinib + corticosteroids vs corticosteroids alone | JAK1/2 inhibitor | Any donor type Ages 14-65 years | Recruiting | Day-28 ORR |
| NCT04167514 | High-risk aGVHD requiring corticosteroids (first-line therapy) | Randomized, double-blind, placebo-controlled, multicenter phase 3 AAT + corticosteroids vs corticosteroids alone | Serine protease inhibitor | Any donor type, conditioning Ages ≥18 years | Recruiting | Day-28 CR, PR |
| NCT04291261 | High-risk aGVHD, AA score 2-3 | Single-arm phase 2 ECP with methoxsalen +2 mg/kg corticosteroids | Induces apoptosis of mononuclear cells | Any donor type, conditioning Ages ≥18 years | Active, not recruiting | Day-28 CR |
| NCT04397367 | Grade 2-4 or high-risk aGVHD (based on ST2, REG3α, or other experiment object) | Nonrandomized Ruxolitinib + corticosteroids | JAK1/2 inhibitor | Any donor type Ages 14-65 years | Recruiting | Day-28 CR |
| Study number | GVHD risk category | Study type/intervention | Treatment target | Transplant eligibility criteria | Study status | Primary outcome measures |
|---|---|---|---|---|---|---|
| Standard-risk aGVHD | ||||||
| NCT02806947 BMT CTN 1501(19) | Standard-risk (Minnesota risk), previously untreated aGVHD requiring systemic therapy, AA score 1-2 biomarker assessment (performed at randomization) | Randomized, multicenter, phase 2 Sirolimus vs prednisone 2 mg/kg | mTOR inhibitor | Any donor type, conditioning Child, adult, older-adult ages | Completed | Day-28 CR/PR rates similar for sirolimus vs prednisone (64.8% vs 73%) Sirolimus associated with reduced steroid exposure and hyperglycemia, improved QOL, and increased risk for TMA |
| NCT03846479 | Low-risk aGVHD, Minnesota standard risk, AA score 1 | Single arm, monotherapy Itacitinib | JAK1 inhibitor | Any donor type, conditioning Ages 12-75 years | Recruiting | Day-28 Minnesota standard-risk clinical criteria (CR, PR, TRM) Day-28 AA score 1 |
| NCT04144036 | Standard-risk aGVHD, Minnesota risk, AA score 1-2 | Single-center nonrandomized phase 1 Neihulizumab dose escalation | Humanized monoclonal antibody binding to CD162 (PSGL-1) | Any donor type, conditioning Ages ≥18 years | Recruiting | Day-28 SAE, MTD of neihulizumab, plasma Cmax |
| High-risk aGVHD | ||||||
| NCT02133924 | High-risk aGVHD, AA score 3 | Multicenter, phase 2 Natalizumab + prednisone | Humanized recombinant antibody binding to alpha-4 integrin | Any donor type, conditioning Ages ≥18 years | Recruiting | Day-28 CR |
| NCT0252502917 | Arm 1: high-risk aGVHD (Minnesota risk, or AA score 3, or AREG ≥33 pg/mL) Arm 2a: steroid-dependent aGVHD Arm 2b: SR-aGVHD | Nonrandomized phase ½ Standard of care IST + Pregnyl | uhCG, which may promote immune tolerance, provides source of EGF for epithelial repair (Pregnyl®) | Any donor type, conditioning Ages 0-76 years | Active, not recruiting | Phase 1: MTD of uhCG/EGF in combination with standard IST Phase 2: day-28 CR, PR, MR, and no response |
| NCT03158896 | De novo high-risk aGVHD SR-aGVHD | Phase 1, dose escalation Umbilical cord-derived, ex vivo cultured, and expanded Wharton's Jelly MSC | Immunosuppressive properties: suppress proliferation of activated T cells, increase production of regulatory T cells, shift immune response toward tolerance | Ages 18-75 years | Active, not recruiting | Day-45 treatment-related SAE |
| NCT04061876 | High-risk grade 2-4 aGVHD (based on ST2, REG3α, or other experiment object) | Randomized, phase 2 Ruxolitinib + corticosteroids vs corticosteroids alone | JAK1/2 inhibitor | Any donor type Ages 14-65 years | Recruiting | Day-28 ORR |
| NCT04167514 | High-risk aGVHD requiring corticosteroids (first-line therapy) | Randomized, double-blind, placebo-controlled, multicenter phase 3 AAT + corticosteroids vs corticosteroids alone | Serine protease inhibitor | Any donor type, conditioning Ages ≥18 years | Recruiting | Day-28 CR, PR |
| NCT04291261 | High-risk aGVHD, AA score 2-3 | Single-arm phase 2 ECP with methoxsalen +2 mg/kg corticosteroids | Induces apoptosis of mononuclear cells | Any donor type, conditioning Ages ≥18 years | Active, not recruiting | Day-28 CR |
| NCT04397367 | Grade 2-4 or high-risk aGVHD (based on ST2, REG3α, or other experiment object) | Nonrandomized Ruxolitinib + corticosteroids | JAK1/2 inhibitor | Any donor type Ages 14-65 years | Recruiting | Day-28 CR |
AAT, alpha-1 antitrypsin; ECP, extracorporeal photopheresis; IST, immunosuppressive therapy; JAK1, Janus kinase 1; MR, mixed response; MSC, mesenchymal stem cell; MTD, maximum tolerated dose; mTOR, mechanistic target of rapamycin; PR, partial response; PSGL-1, P-selectin glycoprotein 1; QOL, quality of life; REG3α, regenerating islet-derived 3-alpha; SAE, serious adverse event; ST2, suppressor of tumorigenesis; TMA, thrombotic microangiopathy; TRM, transplant-related mortality.
CLINICAL CASE
A 61-year-old woman with a history of acute myeloid leukemia in second complete remission underwent fludarabine and melphalan conditioned matched unrelated (male) donor peripheral blood stem cell transplant. She received GVHD prophylaxis with tacrolimus and methotrexate. Her transplant course was complicated by neutropenic fever, protein/calorie malnutrition requiring total parenteral nutrition (TPN), and grade 1 mucositis. On day 11 she developed a maculopapular skin rash involving 36% BSA, and her antibiotics were changed due to concern about a possible drug reaction. On day 14 she developed increasing diarrhea with a negative infectious workup. On day 23, she had a rash with 30% BSA involvement, nausea, emesis, and diarrhea (<1500 mL/d). She was diagnosed with stage 2 skin, stage 1 upper-GI, stage 2 lower-GI, and overall grade 3 Minnesota high-risk aGVHD and started on 2 mg/kg/d methylprednisolone. Her skin rash improved; however, she continued to have nausea and watery diarrhea (1000-1500 mL/d) progressing to ileus. Her hospital course was also complicated by cytomegalovirus reactivation treated with ganciclovir, a vancomycin-resistant Enterococcus faecium urinary tract infection treated with linezolid, and BK cystitis palliated with pyridium. The plan was to start ruxolitinib after insurance approval and with the control of cytomegalovirus reactivation; however, she was never able to start therapy. On day 32 post-HCT, she developed a productive cough, hypotension, and tachycardia and required oxygen and was transferred to the intensive care unit for the management of septic shock. Labs showed pancytopenia and new lung infiltrates. She was given high-flow oxygen, norepinephrine, and broad-spectrum antibiotics. There was some initial improvement, but on day 36 she developed atrial fibrillation with rapid ventricular response, was started on an amiodarone drip, and developed respiratory distress. Her respiratory status worsened, and she died on day 41.
SR-aGVHD
Definition of SR-aGVHD
The EBMT-NIH-CIBMTR Task Force suggested the following definitions for SR-aGVHD or steroid-resistant aGVHD:1 progression of aGVHD within 3 to 5 days of treatment with ≥2 mg/kg/d prednisone equivalent, or2 failure to improve with 5 to 7 days of treatment, or incomplete response after more than 28 days of immunosuppressive therapy including steroids.3,11 SR-aGVHD has also been recognized as (a) worsening GVHD manifestations in patients receiving ≥1 mg/kg/d prednisone equivalent ≥2 days prior to steroid dose tapering; (b) persistent grade 2 to 4 GVHD without improvement ≥7 days during continued treatment with >0.4 mg/kg/d prednisone equivalent, or (c) initial improvement followed by exacerbation ≥3 days during steroid taper at any dose of >0.4 mg/kg/d prednisone equivalent.20 In practice, it can be very challenging to know how long to wait before adding a second-line agent.
Treatment options for SR-aGVHD
In May 2019, the FDA approved the first treatment for SR-aGVHD: ruxolitinib, an inhibitor of Janus kinase 1 and 2, for pediatric and adult patients 12 years of age or older.21 FDA approval was based upon the REACH-1 trial (NCT02953678, study INCB 18424-271), a phase 2, multicenter, open-label, single-arm study of 71 patients, 49 of whom had grades 2 to 4 SR-aGVHD (including patients who failed steroid treatment with or without receiving additional GVHD therapy).21 A starting dose of ruxolitinib, 5 mg twice daily, was administered with methylprednisolone.5 At day 28, the overall response rate (ORR) was 55% (83% for grade 2, 41% for grade 3, and 43% for grade 4 aGVHD), including 27% with a CR, with a median time to first response of 7 days (range, 6-49). Ruxolitinib was further investigated in a phase 3, multicenter, open-label randomized trial comparing the efficacy of ruxolitinib vs investigator's choice of therapy in patients 12 years of age and older with SR-aGVHD (REACH-2, NCT02913261).22 Day-28 ORR was higher in the ruxolitinib group vs the control group (62% vs 39%, P < .001), with 34% and 19% CR, respectively; durable day-56 ORR was also higher in the ruxolitinib group vs the control group (40% vs 22%, P < .001). Ruxolitinib was associated with a higher incidence of thrombocytopenia and a modest increase in anemia and cytomegalovirus infection. In the REACH-1 study, the median time to death or new aGVHD therapy was 5.7 months; in the REACH-2 study, median failure-free survival was 5.0 months.21,22 Previous series have shown that most patients die from SR-aGVHD within 6 months of diagnosis, and only 25% to 30% of patients survive beyond 2 years.23,24
Other experimental approaches to SR-aGVHD with varying mechanisms of action are being tested in clinical trials, recently summarized by Abedin and Hamadani and Martin.20,25 With admirable humility in his “How I Treat” publication, Dr Martin admitted, “Truth be told, I do not know how to treat SR-aGVHD.” This statement underscores the tremendous difficulty in overcoming this largely fatal disease despite decades of intense study. Truth be told, we do not fully understand the pathophysiology of SR-aGVHD. Most novel therapeutics have been potent immunosuppressants, yet that may not be the correct or only approach. In a preclinical model, there were no differences in T-cell expansion, activation, or enhanced cytokine production in donor T cells in mice with responsive vs refractory aGVHD.26 Recently, murine SR-aGVHD was described as driven by interleukin 22-dependent dysbiosis, with a protective role of CXCR3hi mononuclear phagocytes in reducing bacterial translocation.27 Gene expression studies from human colorectal biopsies showed that human SR-aGVHD is characterized by tissue response to damage, cellular stress, and macrophage accumulation, not T-cell proliferation.28 Collectively, these recent studies suggest that future therapeutic efforts in SR-aGVHD, in addition to targeting the initial T-cell-mediated damage and inflammation, might also consider studies of agents designed to enhance tissue repair and to correct dysbiosis while trying to avoid broad immunosuppression and its inherent risks of infection17,29-34 . Recently described targets such as CD83 suggest this may be feasible.35
CLINICAL CASE
A 23-year-old man underwent a myeloablative, matched unrelated donor peripheral blood stem cell transplant for high-risk T-cell acute lymphoblastic leukemia and was diagnosed with stage 2 skin, stage 3 GI, overall Minnesota high-risk aGVHD. He was treated with high-dose steroids, urinary-derived human chorionic gonadotropin (uhCG), and EGF in clinical trial NCT02525029. He achieved a CR at 4 weeks after the initiation of therapy, which was sustained at 8 weeks. Over the ensuing 2 years, he had 6 hospitalizations with a recurrence of nausea, vomiting, and diarrhea. With each episode he was tested for infections (1 episode was due to norovirus) and other complications, yet he would transiently improve with a modest increase in prednisone dose. At 3 years posttransplant, he was readmitted with a body mass index of 16 and recalcitrant anorexia and diarrhea. Repeat intestinal biopsies showed villous blunting, but the patient and donor absence of HLA-DQ2/8 ruled out celiac disease. Fecal elastase was mildly low at 168 µg/g of stool, suggestive of mild/moderate pancreatic exocrine insufficiency. Fecal calprotectin was mildly elevated at 72.8 mg/kg, suggesting the presence of inflammation/neutrophils in the intestinal lumen (normal, 0-49.9 mg/kg). Plasma AREG was mildly elevated at 29.1 pg/mL, suggesting tissue damage and mild systemic inflammation (normal, <5 pg/mL; active severe intestinal aGVHD is typically >33 pg/ml). Plasma citrulline was low at 0.9 µmol/dL (normal, 1.3-6.0 µmol/dL; values <1 µmol/dL are predictive of total villous atrophy).36 D-xylose absorption was low, with a serum level of 7 pg/mL at 2 hours (normal, 32-58 pg/mL), suggesting malabsorption. Budesonide and TPN were resumed. After a weeklong hospitalization, he was discharged from the hospital tolerating some oral intake but likely to be dependent on TPN long-term given the findings of low enterocyte mass (low citrulline) and malabsorption (low D-xylose absorption).
Steroid-dependent aGVHD
This patient highlights a common clinical scenario of a prolonged aGVHD course of waxing and waning symptoms. “Steroid-dependent” aGVHD can be defined as (a) only achieving a partial (not complete) response to steroids after 8 weeks, (b) still requiring >10 mg/m2 prednisone after 8 weeks or any prednisone at all after 10 weeks, or (c) a flare of aGVHD symptoms requiring at least a 25% increase in prednisone dose.37 Steroid-dependent aGVHD is experienced by 31% of patients with aGVHD.37 While it is not associated with increased mortality, it may be associated with morbidity and a prolonged health care burden, as this case description highlights. This patient had high-risk aGVHD with an initial response to high-dose corticosteroids and uhCG/EGF but subsequently flared, suggesting that one or more underlying causes had not been completely addressed by his initial therapy.
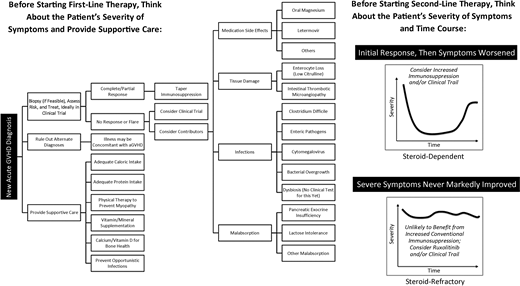
Figure 1 shows the differential diagnoses and considerations in evaluating aGVHD. An increase in immunosuppression may not be the right treatment for all patients with worsening symptoms. Several questions must be answered in the coming years to improve outcomes. Can biomarkers be used repeatedly over weeks to months as a guide to tapering immunosuppression? Which patients need different modes of supportive care (eg, remediation of dysbiosis vs tissue damage), and can this even be distinguished biologically? How long should adjunct repair-based therapies such as uhCG/EGF be continued to achieve maximal mucosal healing? What other targets of aGVHD (eg, the endothelium) should be treated? Additional clinical trials are urgently needed to address these questions.
Conflict-of-interest disclosure
Shernan G. Holtan: advisor: Incyte, Generon.
Laura F. Newell: no conflicts to disclose.
Off-label drug use
Laura F. Newell: no off-label drug use.
Shernan G. Holtan: Off-label drug use for SGH: Urinary-derived human chorionic gonadotropin has Orphan Drug Designation from the Food and Drug Administration for the treatment of acute GVHD.
